
CiberAMP: An R Package to Identify Differential mRNA Expression Linked to Somatic Copy Number Variations in Cancer Datasets
 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. TCGA RNA-Seq Data Download, Filtering, and Normalization
2.2. Differential Expression Analysis
2.3. Classification of Tumor Samples
2.4. Copy Number and mRNA Expression Data Integration
2.5. Analysis of Co-Amplified and Co-Deleted Genes
2.6. Identification of SCNV-DEG Enriched Genomic Regions
2.7. Classification Algorithm
2.8. Data Visualization
2.9. Analysis of the Genetic Dependence of Cancer Cell Lines on Gene Amplification
3. Results and Discussion
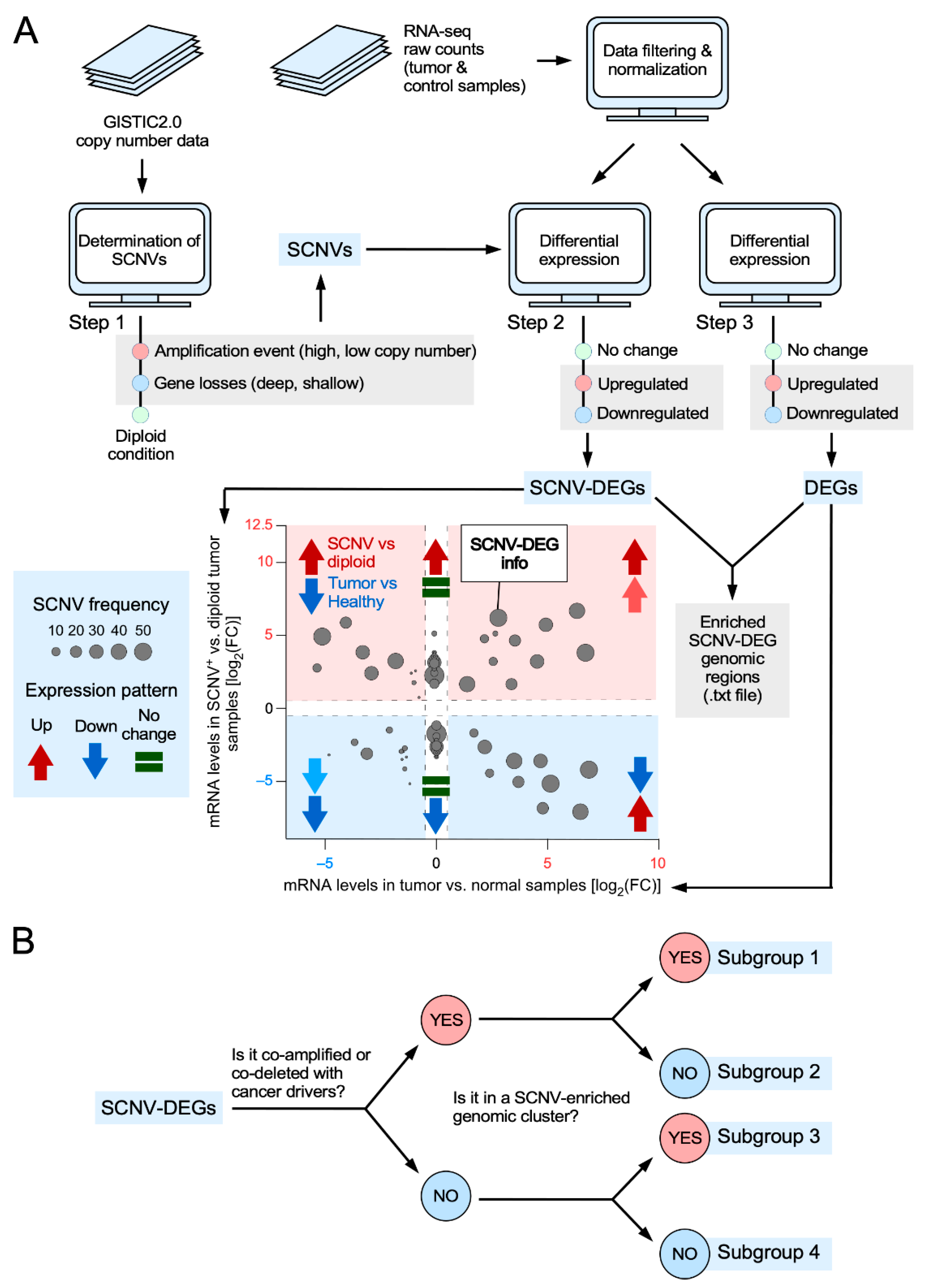
3.1. Features of the CiberAMP Package
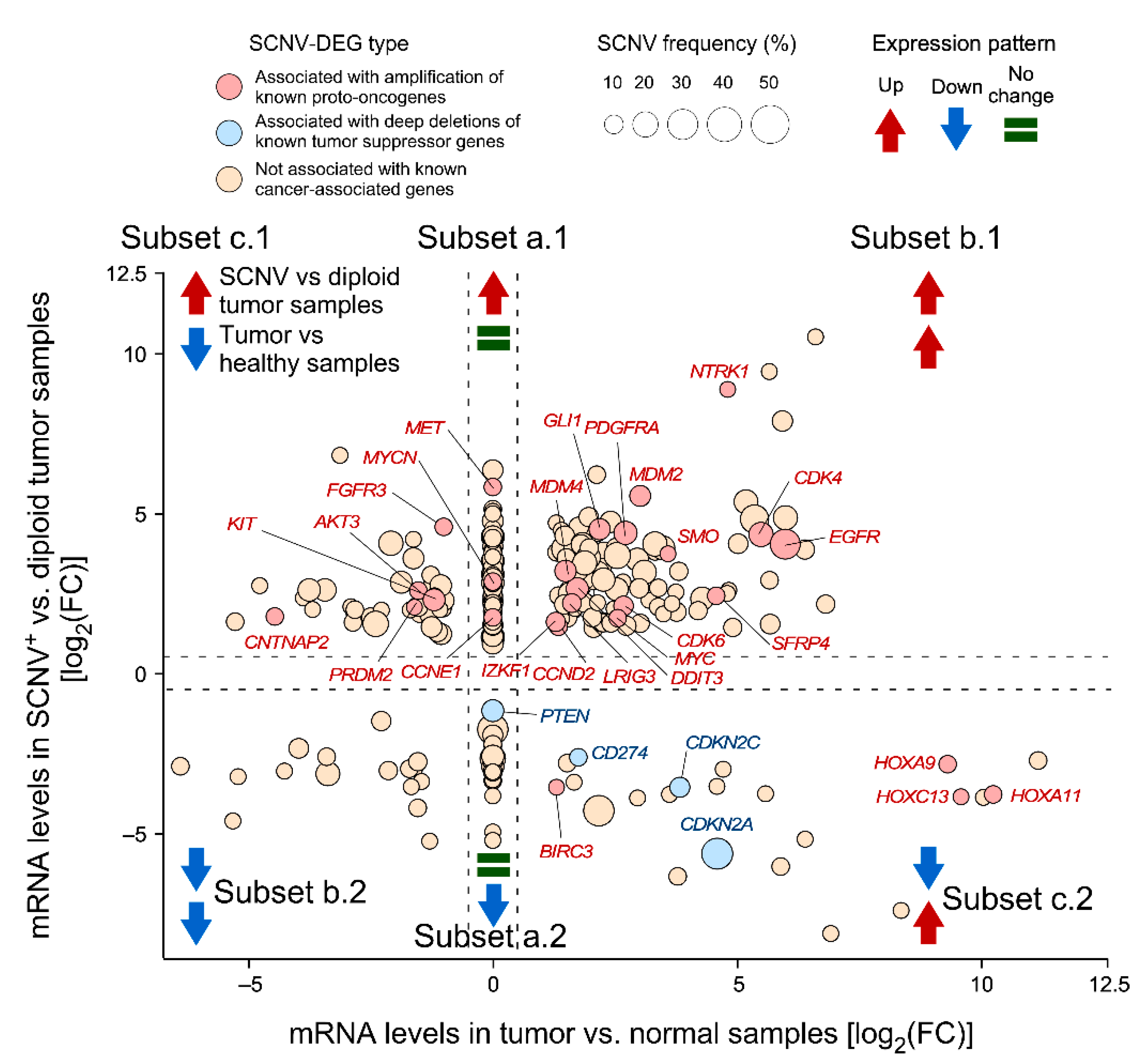
3.2. Identification of SCNV-DEGs in Glioblastoma Using CiberAMP
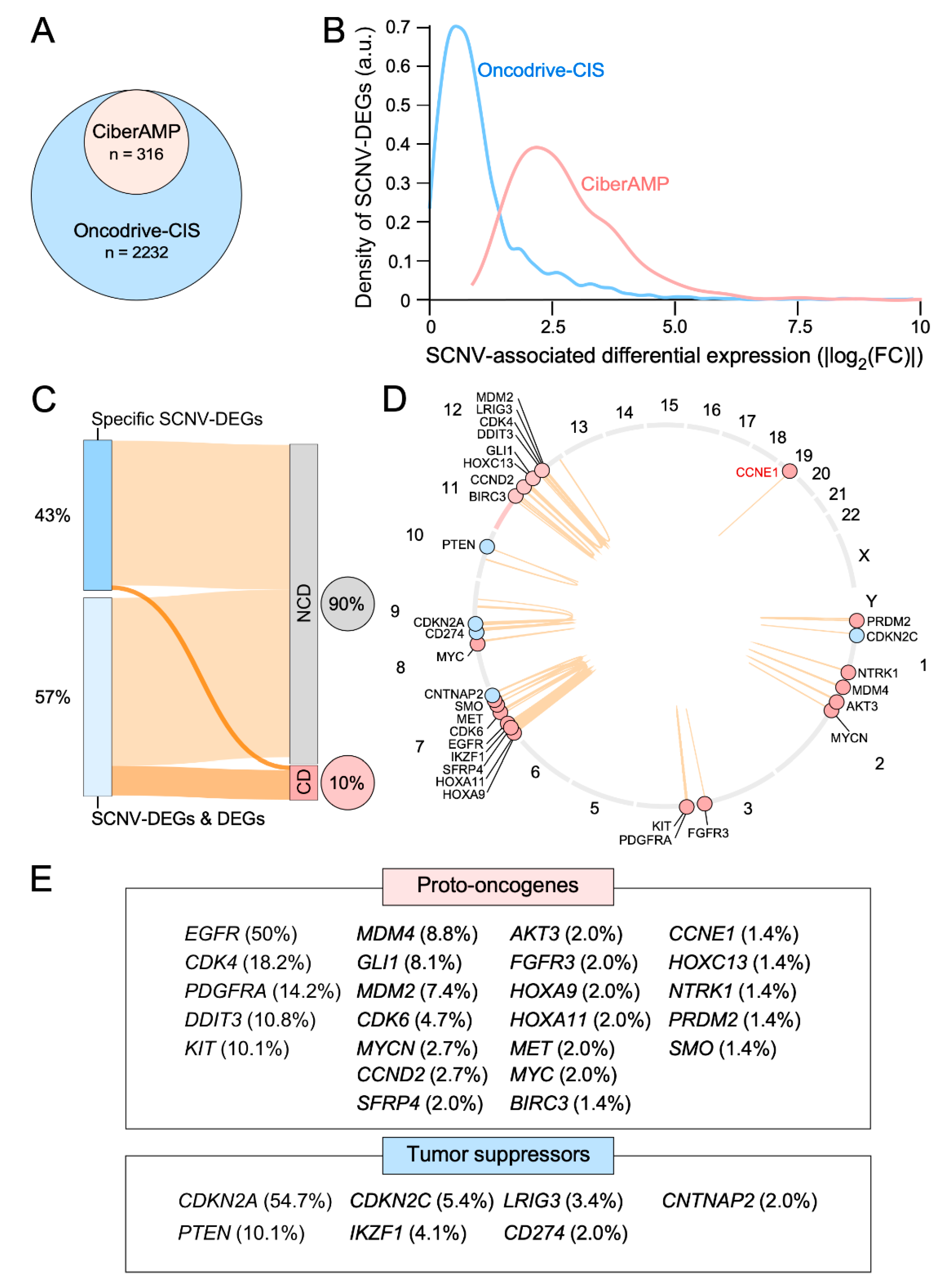
3.3. CiberAMP Benchmarking
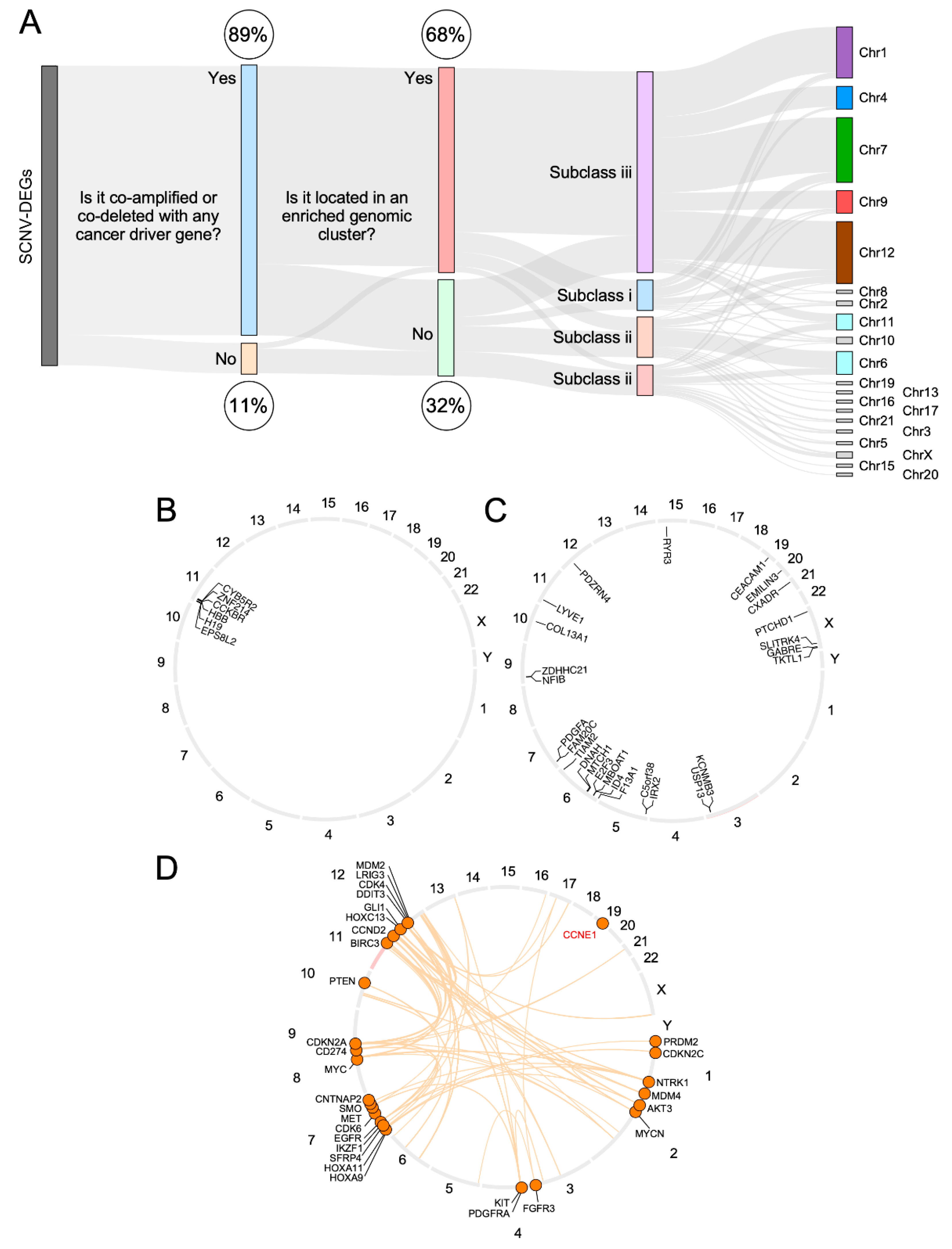
3.4. CiberAMP Provides Information for Subsequent Hypothesis-Driven Studies on SCNV-DEGs
3.5. Validation of CiberAMP-Identified SCNV-DEGs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Henrichsen, C.N.; Chaignat, E.; Reymond, A. Copy number variants, diseases and gene expression. Hum. Mol. Genet. 2009, 18, R1–R8. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Anastassiou, D. Pan-cancer driver copy number alterations identified by joint expression/CNA data analysis. Sci. Rep. 2020, 10, 17199. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.-Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Lv, N.; Liao, J.; Long, J.; Xue, R.; Ai, N.; Xu, D.; Fan, X. Copy number variation is highly correlated with differential gene expression: A pan-cancer study. BMC Med. Genet. 2019, 20, 75. [Google Scholar] [CrossRef]
- Disel, U.; Madison, R.; Abhishek, K.; Chung, J.H.; Trabucco, S.E.; Matos, A.O.; Frampton, G.M.; Albacker, L.A.; Reddy, V.; Karadurmus, N.; et al. The Pan-Cancer Landscape of Coamplification of the Tyrosine Kinases KIT, KDR, and PDGFRA. Oncologist 2020, 25, e39–e47. [Google Scholar] [CrossRef]
- Hyman, E.; Kauraniemi, P.; Hautaniemi, S.; Wolf, M.; Mousses, S.; Rozenblum, E.; Ringnér, M.; Sauter, G.; Monni, O.; Elkahloun, A.; et al. Impact of DNA amplification on gene expression patterns in breast cancer. Cancer Res. 2002, 62, 6240–6245. [Google Scholar]
- Pollack, J.R.; Sorlie, T.; Perou, C.M.; Rees, C.A.; Jeffrey, S.S.; Lonning, P.E.; Tibshirani, R.; Botstein, D.; Borresen-Dale, A.-L.; Brown, P.O. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 12963–12968. [Google Scholar] [CrossRef]
- Bicciato, S.; Spinelli, R.; Zampieri, M.; Mangano, E.; Ferrari, F.; Beltrame, L.; Cifola, I.; Peano, C.; Solari, A.; Battaglia, C. A computational procedure to identify significant overlap of differentially expressed and genomic imbalanced regions in cancer datasets. Nucleic Acids Res. 2009, 37, 5057–5070. [Google Scholar] [CrossRef]
- Chin, K.; Devries, S.; Fridlyand, J.; Spellman, P.T.; Roydasgupta, R.; Kuo, W.-L.; Lapuk, A.; Neve, R.M.; Qian, Z.; Ryder, T.; et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006, 10, 529–541. [Google Scholar] [CrossRef]
- Lee, H.; Kong, S.W.; Park, P.J. Integrative analysis reveals the direct and indirect interactions between DNA copy number aberrations and gene expression changes. Bioinformatics 2008, 24, 889–896. [Google Scholar] [CrossRef]
- Salari, K.; Tibshirani, R.; Pollack, J.R. DR-Integrator: A new analytic tool for integrating DNA copy number and gene expression data. Bioinformatics 2010, 26, 414–416. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, M.; Schwender, H.; Merk, S.; Haferlach, C.; Ickstadt, K.; Dugas, M. Integrated analysis of copy number alterations and gene expression: A bivariate assessment of equally directed abnormalities. Bioinformatics 2009, 25, 3228–3235. [Google Scholar] [CrossRef]
- Van Wieringen, W.N.; Van De Wiel, M.A. Nonparametric Testing for DNA Copy Number Induced Differential mRNA Gene Expression. Biometrics 2009, 65, 19–29. [Google Scholar] [CrossRef]
- Choi, H.; Qin, Z.S.; Ghosh, D. A Double-Layered Mixture Model for the Joint Analysis of DNA Copy Number and Gene Expression Data. J. Comput. Biol. 2010, 17, 121–137. [Google Scholar] [CrossRef]
- Menezes, R.X.; Boetzer, M.; Sieswerda, M.; Van Ommen, G.-J.B.; Boer, J.M. Integrated analysis of DNA copy number and gene expression microarray data using gene sets. BMC Bioinform. 2009, 10, 203. [Google Scholar] [CrossRef]
- Lê Cao, K.-A.; González, I.; Déjean, S. integrOmics: An R package to unravel relationships between two omics datasets. Bioinformatics 2009, 25, 2855–2856. [Google Scholar] [CrossRef]
- Louhimo, R.; Lepikhova, T.; Monni, O.; Hautaniemi, S. Comparative analysis of algorithms for integration of copy number and expression data. Nat. Methods 2012, 9, 351–355. [Google Scholar] [CrossRef]
- Lahti, L.; Schafer, M.; Klein, H.-U.; Bicciato, S.; Dugas, M. Cancer gene prioritization by integrative analysis of mRNA expression and DNA copy number data: A comparative review. Brief. Bioinform. 2013, 14, 27–35. [Google Scholar] [CrossRef]
- Karlsson, J.; Larsson, E. FocalScan: Scanning for altered genes in cancer based on coordinated DNA and RNA change. Nucleic Acids Res. 2016, 44, e150. [Google Scholar] [CrossRef]
- Tamborero, D.; Lopez-Bigas, N.; Gonzalez-Perez, A. Oncodrive-CIS: A Method to Reveal Likely Driver Genes Based on the Impact of Their Copy Number Changes on Expression. PLoS ONE 2013, 8, e55489. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Risso, D.; Schwartz, K.; Sherlock, G.; Dudoit, S. GC-Content Normalization for RNA-Seq Data. BMC Bioinform. 2011, 12, 480. [Google Scholar] [CrossRef]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Corchete, L.A.; Rojas, E.A.; Alonso-López, D.; De Las Rivas, J.; Gutiérrez, N.C.; Burguillo, F.J. Systematic comparison and assessment of RNA-seq procedures for gene expression quantitative analysis. Sci. Rep. 2020, 10, 19737. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; Mcfarland, J.M. Chronos: A cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 2021, 22, 343. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Ming, H.; Lan, Y.; He, F.; Xiao, X.; Zhou, X.; Zhang, Z.; Li, P.; Huang, G. Cytochrome b5 reductase 2 suppresses tumor formation in nasopharyngeal carcinoma by attenuating angiogenesis. Chin. J. Cancer 2015, 34, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.S.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Intratumoral Heterogeneity for Inactivating Frameshift Mutation of CYB5R2 Gene in Colorectal Cancers. Pathol. Oncol. Res. 2020, 26, 585–586. [Google Scholar] [CrossRef]
- Meng, L.L.; Wang, J.L.; Xu, S.P.; Zu, L.D.; Yan, Z.W.; Zhang, J.B.; Han, Y.Q.; Fu, G.H. Low serum gastrin associated with ER. BMC Cancer 2018, 18, 824. [Google Scholar] [CrossRef]
- Zhang, R.; Li, M.; Zang, W.; Chen, X.; Wang, Y.; Li, P.; Du, Y.; Zhao, G.; Li, L. MiR-148a regulates the growth and apoptosis in pancreatic cancer by targeting CCKBR and Bcl-2. Tumour. Biol. 2014, 35, 837–844. [Google Scholar] [CrossRef]
- Cui, Y.; Li, S.B.; Peng, X.C.; Wu, J.; Fu, G.H. Trastuzumab Inhibits Growth of HER2-Negative Gastric Cancer Cells Through Gastrin-Initialized CCKBR Signaling. Dig. Dis. Sci. 2015, 60, 3631–3641. [Google Scholar] [CrossRef]
- Hayashi, Y.; Ueki, K.; Waha, A.; Wiestler, O.D.; Louis, D.N.; Deimlina, A. Association of EGFR Gene Amplification and CDKN2 (p16/MTS1) Gene Deletion in Glioblastoma Multiforme. Brain Pathol. 1997, 7, 871–875. [Google Scholar] [CrossRef]
- Ma, S.; Rudra, S.; Campian, J.L.; Dahiya, S.; Dunn, G.P.; Johanns, T.; Goldstein, M.; Kim, A.H.; Huang, J. Prognostic impact of CDKN2A/B deletion, TERT mutation, and EGFR amplification on histological and molecular IDH-wildtype glioblastoma. Neuro-Oncol. Adv. 2020, 2, vdaa126. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Xu, L.; Zhang, J.; Cao, H. Analysis of the EGFR Amplification and CDKN2A Deletion Regulated Transcriptomic Signatures Reveals the Prognostic Significance of. Front. Oncol. 2021, 11, 551160. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | CiberAMP | Oncodrive-CIS |
|---|---|---|
| Language | R | Python |
| Input data | List of gene symbols and TCGA cohort IDs | Normalized RNA-seq and copy number data |
| Samples from healthy tissue | Yes | Yes |
| Score provided | Log2(FC) and adjusted p value | Combined score and adjusted p value |
| Use of validated pipelines for differential expression analyses | Yes | No |
| Provides log2(FC) values associated with the differential expression analyses | Yes | No |
| Provides information on differential gene expression between copy number altered and diploid tumor samples | Yes | No |
| Can analyze different tumor types in the same run | Yes | No |
| Provides information on concurrency or mutual exclusivity among SCNV-DEGs | Yes | No |
| Provides information on enriched SCNV-DEG genomic regions | Yes | No |
| Includes an interactive visualization tool to explore the outputs obtained from the analyses | Yes | No |
| Bin | CiberAMP | Oncodrive-CIS | ||
|---|---|---|---|---|
| # Cancer Drivers | % Cancer Drivers | # Cancer Drivers | % Cancer Drivers | |
| Glioblastoma | ||||
| 0–10% | 4 | 12.9 | 16 | 7.2 |
| 10–20% | 10 | 15.9 | 33 | 7.4 |
| 20–30% | 12 | 12.8 | 43 | 6.4 |
| 30–40% | 15 | 11.9 | 50 | 5.6 |
| 40–50% | 19 | 12.0 | 61 | 5.5 |
| 50–60% | 22 | 11.6 | 71 | 5.3 |
| 60–70% | 26 | 11.8 | 80 | 5.1 |
| 70–80% | 27 | 10.7 | 92 | 5.1 |
| 80–90% | 29 | 10.2 | 108 | 0.5 |
| 90–100% | 31 (total) | 9.8 | 119 (total) | 5.3 |
| Head and neck cancer | ||||
| 0–10% | 16 | 8.0 | 68 | 5.1 |
| 10–20% | 29 | 7.3 | 120 | 4.5 |
| 20–30% | 38 | 6.4 | 163 | 4.1 |
| 30–40% | 44 | 5.5 | 219 | 4.1 |
| 40–50% | 49 | 4.9 | 259 | 3.9 |
| 50–60% | 53 | 4.4 | 315 | 3.9 |
| 60–70% | 61 | 4.4 | 369 | 3,9 |
| 70–80% | 65 | 4.1 | 424 | 3.9 |
| 80–90% | 69 | 3.8 | 467 | 3.9 |
| 90–100% | 76 (total) | 3.8 | 514 (total) | 3.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caloto, R.; Lorenzo-Martín, L.F.; Quesada, V.; Carracedo, A.; Bustelo, X.R. CiberAMP: An R Package to Identify Differential mRNA Expression Linked to Somatic Copy Number Variations in Cancer Datasets. Biology 2022, 11, 1411. https://doi.org/10.3390/biology11101411
Caloto R, Lorenzo-Martín LF, Quesada V, Carracedo A, Bustelo XR. CiberAMP: An R Package to Identify Differential mRNA Expression Linked to Somatic Copy Number Variations in Cancer Datasets. Biology. 2022; 11(10):1411. https://doi.org/10.3390/biology11101411
Chicago/Turabian StyleCaloto, Rubén, L. Francisco Lorenzo-Martín, Víctor Quesada, Arkaitz Carracedo, and Xosé R. Bustelo. 2022. "CiberAMP: An R Package to Identify Differential mRNA Expression Linked to Somatic Copy Number Variations in Cancer Datasets" Biology 11, no. 10: 1411. https://doi.org/10.3390/biology11101411