
Construction of a High-Density Genetic Linkage Map for the Mapping of QTL Associated with Growth-Related Traits in Sea Cucumber (Apostichopus japonicus)

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mapping Population and DNA Extraction
2.2. GBS Library Construction and Sequencing
2.3. SNP Calling and Genotyping
2.4. Linkage Map Construction
2.5. QTL Mapping and Potential Candidate Gene Identification
2.6. Expression and Growth Association Analyses of a Potential Candidate Gene
3. Results
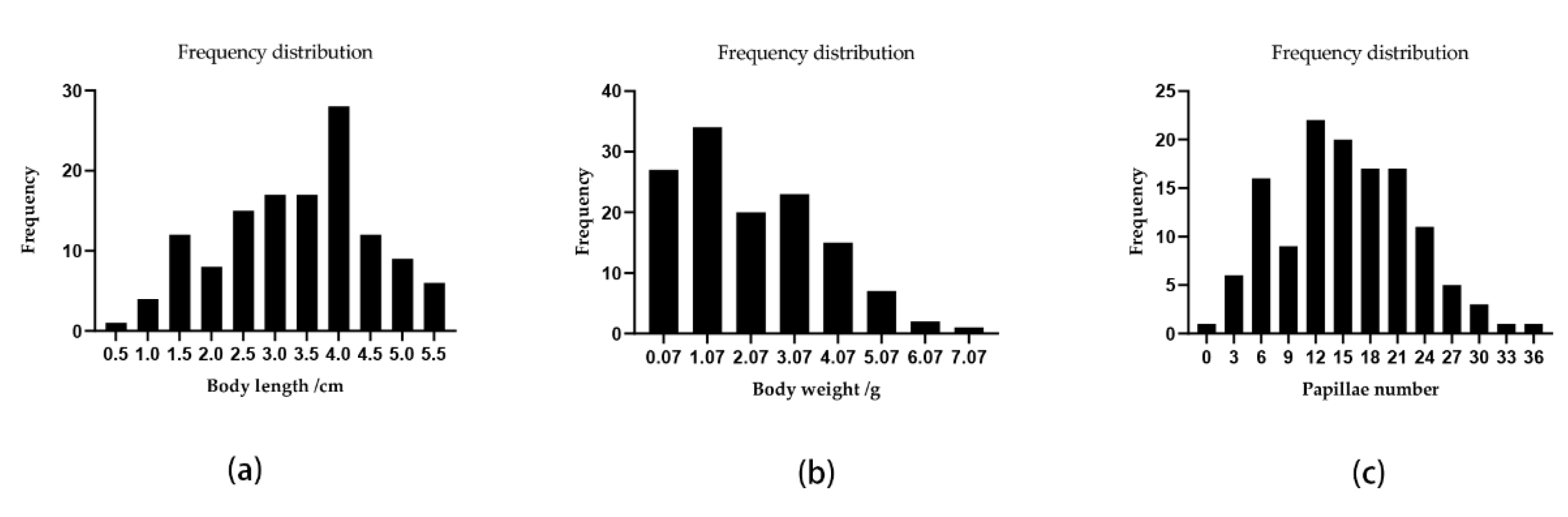
3.1. Genotyping-by-Sequencing and SNP Discovery
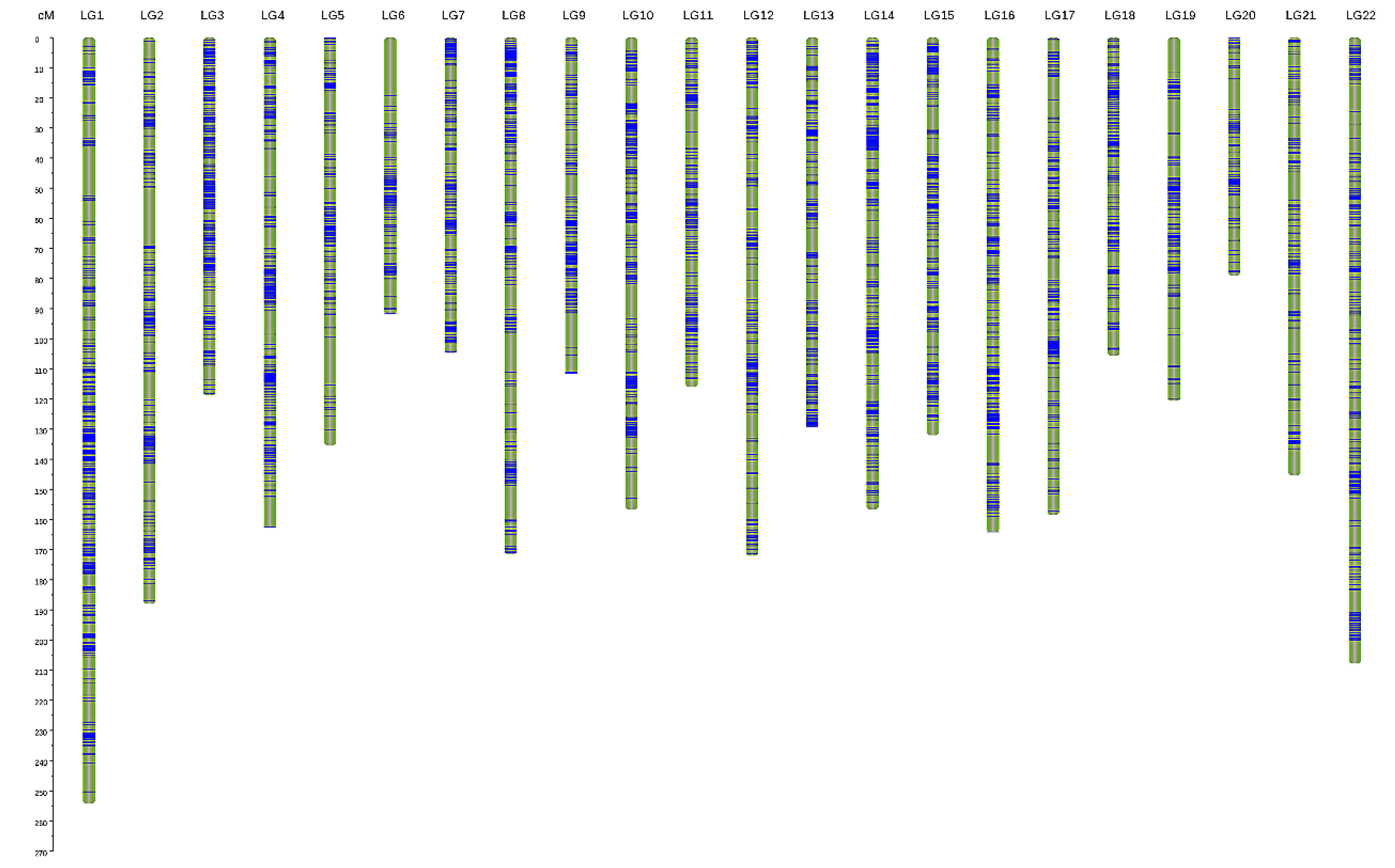
3.2. Genetic Linkage Map Construction
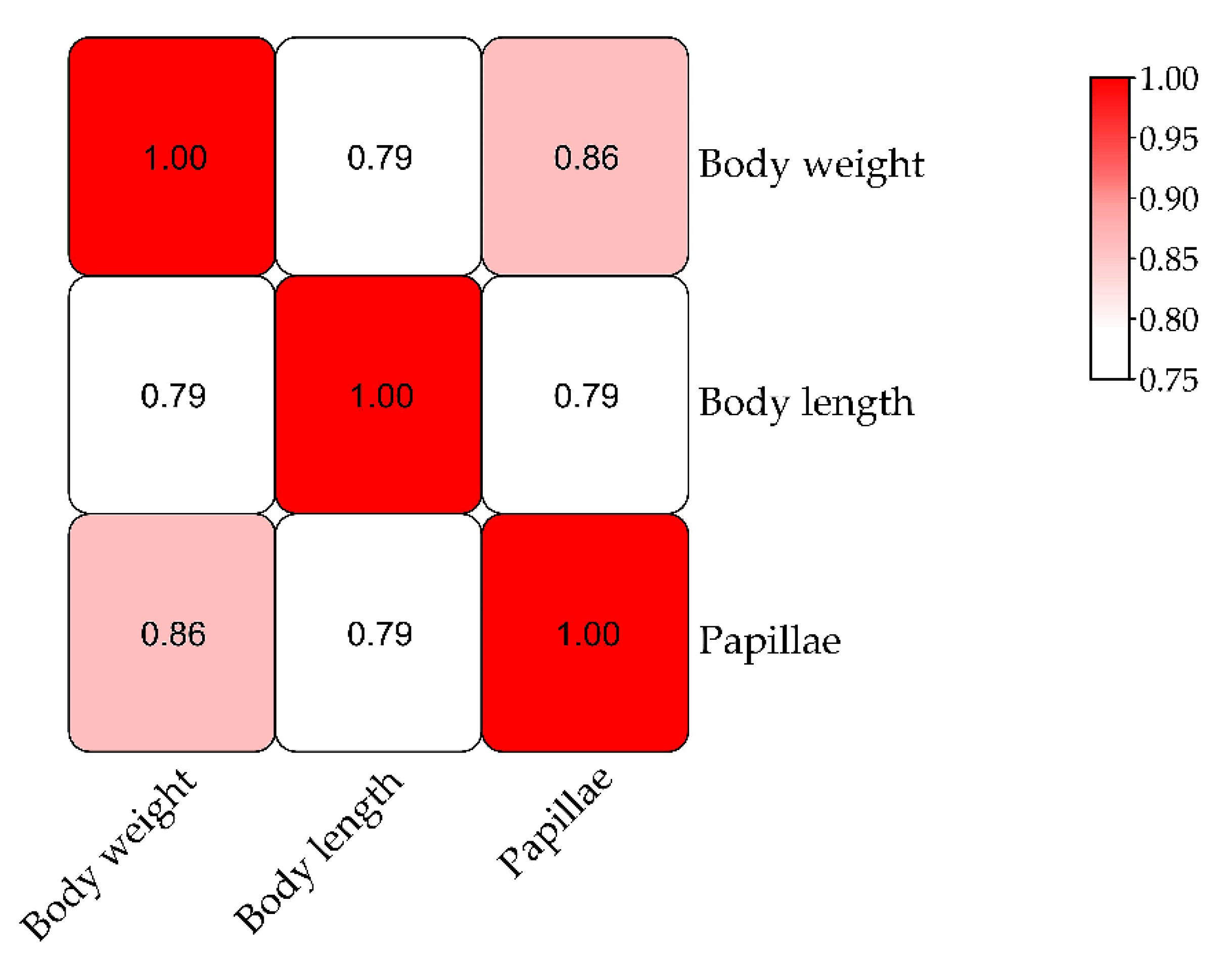
3.3. QTL Mapping and Growth Association Analysis
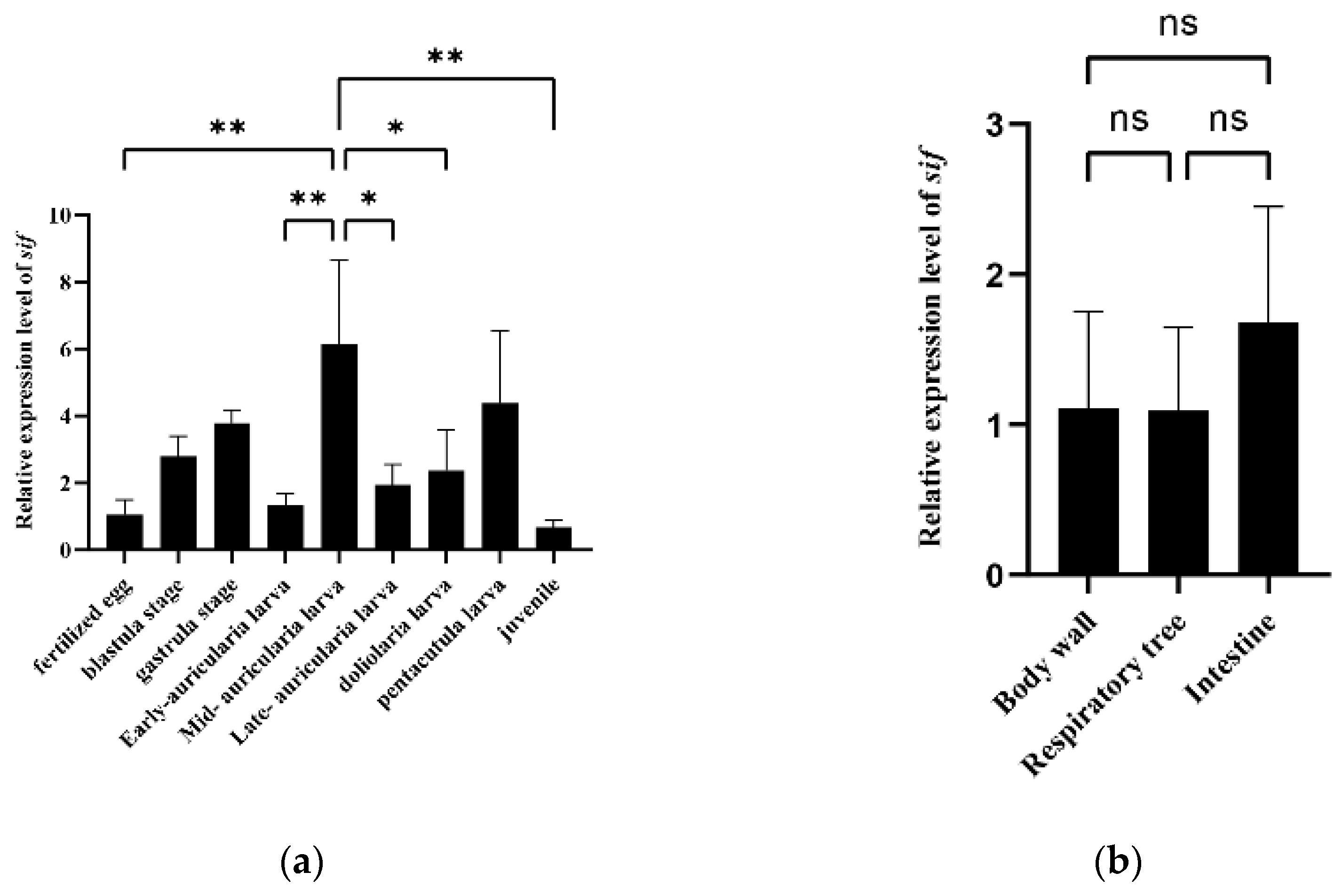
3.4. Expression and Growth Association Analyses of a Potential Candidate Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Okumura, S.-I.; Kimura, K.; Sakai, M.; Waragaya, T.; Furukawa, S.; Takahashi, A.; Yamamori, K. Chromosome number and telomere sequence mapping of the Japanese sea cucumber Apostichopus japonicus. Fish. Sci. 2009, 75, 249–251. [Google Scholar] [CrossRef]
- Yan, J.; Jing, J.; Mu, X.; Du, H.; Tian, M.; Wang, S.; Lu, W.; Bao, Z. A genetic linkage map of the sea cucumber (Apostichopus japonicus) based on microsatellites and SNPs. Aquaculture 2013, 404, 1–7. [Google Scholar] [CrossRef]
- Li, Q.; Chen, L.; Kong, L. A genetic linkage map of the sea cucumber, Apostichopus japonicus (Selenka), based on AFLP and microsatellite markers. Anim. Genet. 2009, 40, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Li, Y.; Jing, J.; Mu, C.; Du, H.; Dou, J.; Mao, J.; Li, X.; Jiao, W.; Wang, Y.; et al. Construction of a high-density genetic map and quantitative trait locus mapping in the sea cucumber Apostichopus japonicus. Sci. Rep. 2015, 5, 14852. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Sun, L.; Yuan, J.; Sun, Y.; Gao, Y.; Zhang, L.; Li, S.; Dai, H.; Hamel, J.F.; Liu, C.; et al. The sea cucumber genome provides insights into morphological evolution and visceral regeneration. PLoS Biol. 2017, 15, e2003790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knapik, E.W.; Goodman, A.; Ekker, M.; Chevrette, M.; Delgado, J.; Neuhauss, S.; Shimoda, N.; Driever, W.; Fishman, M.C.; Jacob, H.J. A microsatellite genetic linkage map for zebrafish (Danio rerio). Nat. Genet. 1998, 18, 338–343. [Google Scholar] [CrossRef]
- Slate, J. Robustness of linkage maps in natural populations: A simulation study. Proc. Biol. Sci. 2008, 275, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Sturtevant, A.H. The linear arrangement of six sex-linked factors in Drosophila, as shown by their mode of association. J. Exp. Zool. 1913, 74, 43–59. [Google Scholar] [CrossRef]
- Kocher, T.; Lee, W.; Sobolewska, H.; Penman, D.J.; Mcandrew, B.J. A genetic linkage map of a cichlid fish, the tilapia (Oreochromis niloticus). Genetics 1998, 148, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Hubert, S.; Hedgecock, D. Linkage maps of microsatellite DNA markers for the pacific oyster Crassostrea gigas. Genetics 2004, 168, 351–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, K.M.; Young, W.P.; Danzmann, R.G.; Robison, B.D.; Rexroad, C.; Noakes, M.; Phillips, R.B.; Bentzen, P.; Spies, I.; Knudsen, K.; et al. A consolidated linkage map for rainbow trout (Oncorhynchus mykiss). Anim. Genet. 2003, 34, 102–115. [Google Scholar] [CrossRef]
- Li, Y.-H.; Wang, H.-P. Advances of genotyping-by-sequencing in fisheries and aquaculture. Rev. Fish Biol. Fish. 2017, 27, 535–559. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.W.; Sambrook, J. Isolation and Quantification of DNA. In Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2001; Volume 1. [Google Scholar]
- Wallace, J.G.; Mitchell, S.E. Genotyping-by-Sequencing. Curr. Protoc. Plant Biol. 2017, 2, 64–77. [Google Scholar]
- Peng, W.; Yu, F.; Wu, Y.; Zhang, Y.; Lu, C.; Wang, Y.; Huang, Z.; Lu, Y.; Chen, N.; Luo, X.; et al. Identification of growth-related SNPs and genes in the genome of the Pacific abalone (Haliotis discus hannai) using GWAS. Aquaculture 2021, 541, 736820. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Van Ooijen, J. JoinMap® 4.0: Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV Wagening. Netherlands. 2006, 33. [Google Scholar] [CrossRef]
- Wu, Y.; Close, T.J.; Lonardi, S. On the accurate construction of consensus genetic maps. Comput. Syst. Bioinform. 2008, 7, 285–296. [Google Scholar]
- Van Ooijen, J. MapQTL® 6, Software for the Mapping of Quantitative Trait Loci in Experimental Populations of Diploid Species; Kyazma BV: Wagening, The Netherlands, 2009; Volume 64. [Google Scholar]
- Li, H. A quick method to calculate QTL confidence interval. J. Genet. 2011, 90, 355–360. [Google Scholar] [CrossRef]
- Li, Y.; Kikuchi, M.; Li, X.; Gao, Q.; Xiong, Z.; Ren, Y.; Zhao, R.; Mao, B.; Kondo, M.; Irie, N.; et al. Weighted gene co-expression network analysis reveals potential genes involved in early metamorphosis process in sea cucumber Apostichopus japonicus. Biochem. Biophys. Res. Commun. 2018, 495, 1395–1402. [Google Scholar] [CrossRef]
- Hu, C.; Xu, Y.; Wen, J.; Zhang, L.; Fan, S.; Su, T. Larval development and juvenile growth of the sea cucumber Stichopus sp. (Curry fish). Aquaculture 2009, 300, 73–79. [Google Scholar] [CrossRef]
- Boyko, A.V.; Girich, A.S.; Eliseikina, M.G.; Maslennikov, S.I.; Dolmatov, I.Y. Reference assembly and gene expression analysis of Apostichopus japonicus larval development. Sci. Rep. 2019, 9, 1131. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Slate, J.; Gratten, J.; Beraldi, D.; Stapley, J.; Hale, M.; Pemberton, J.M. Gene mapping in the wild with SNPs: Guidelines and future directions. Genetica 2009, 136, 97–107. [Google Scholar] [CrossRef]
- Peng, W.; Xu, J.; Zhang, Y.; Feng, J.; Dong, C.; Jiang, L.; Feng, J.; Chen, B.; Gong, Y.; Chen, L.; et al. An ultra-high density linkage map and QTL mapping for sex and growth-related traits of common carp (Cyprinus carpio). Sci. Rep. 2016, 6, 26693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivers, R. Sex differences in rates of recombination and sexual selection. Evol. Sex 1988, 270, 286. [Google Scholar]
- Chang, Y.; Shi, S.; Zhao, C.; Han, Z. Characteristics of papillae in wild, cultivated and hybrid sea cucumbers (Apostichopus japonicus). Afr. J. Biotechnol. 2011, 10, 13780–13788. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, M.; Bordieri, L.; Greenwood, M.P.; Rosso Melo, M.; Colombari, D.S.; Colombari, E.; Paton, J.F.; Murphy, D. Transcription factor CREB3L1 regulates vasopressin gene expression in the rat hypothalamus. J. Neurosci. 2014, 34, 3810–3820. [Google Scholar] [CrossRef] [Green Version]
- Denard, B.; Seemann, J.; Chen, Q.; Gay, A.; Huang, H.; Chen, Y.; Ye, J. The membrane-bound transcription factor CREB3L1 is activated in response to virus infection to inhibit proliferation of virus-infected cells. Cell Host Microbe 2011, 10, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Rudick, M.; Anderson, R.G. Multiple functions of caveolin-1. J. Biol. Chem. 2002, 277, 41295–41298. [Google Scholar] [CrossRef] [Green Version]
- Djakiew, D. Role of nerve growth factor-like protein in the paracrine regulation of prostate growth. J. Androl. 1992, 13, 476–487. [Google Scholar] [PubMed]
- Chen, Y.; Dicou, E.; Djakiew, D. Characterization of nerve growth factor precursor protein expression in rat round spermatids and the trophic effects of nerve growth factor in the maintenance of Sertoli cell viability. Mol. Cell. Endocrinol. 1997, 127, 129–136. [Google Scholar] [CrossRef]
- Hirsch, M.R.; Gaugler, L.; Deagostini-Bazin, H.; Bally-Cuif, L.; Goridis, C. Identification of positive and negative regulatory elements governing cell-type-specific expression of the neural cell adhesion molecule gene. Mol. Cell. Biol. 1990, 10, 1959–1968. [Google Scholar]
- Sone, M.; Hoshino, M.; Suzuki, E.; Kuroda, S.; Kaibuchi, K.; Nakagoshi, H.; Saigo, K.; Nabeshima, Y.; Hama, C. Still life, a protein in synaptic terminals of Drosophila homologous to GDP-GTP exchangers. Neurosci. Res. 1997, 28, 543–547. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LG | Consensus Map | Female Map | Male Map | F:M Distance Ratio (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP Number | Distance (cM) | Average Distance (cM) | SNP Number | Distance (cM) | Average Distance (cM) | SNP Number | Distance (cM) | Average Distance (cM) | ||
| 1 | 522 | 254.19 | 0.487 | 311 | 244.897 | 0.787 | 268 | 116.991 | 0.437 | 2.09 |
| 2 | 282 | 187.96 | 0.667 | 138 | 175.923 | 1.275 | 188 | 93.965 | 0.500 | 1.87 |
| 3 | 400 | 118.72 | 0.297 | 190 | 112.690 | 0.593 | 230 | 75.942 | 0.330 | 1.48 |
| 4 | 339 | 162.74 | 0.480 | 179 | 154.192 | 0.861 | 202 | 90.616 | 0.449 | 1.70 |
| 5 | 194 | 135.31 | 0.697 | 93 | 124.056 | 1.334 | 115 | 71.007 | 0.617 | 1.75 |
| 6 | 134 | 91.77 | 0.685 | 20 | 20.580 | 1.029 | 119 | 91.482 | 0.769 | 0.22 |
| 7 | 261 | 104.68 | 0.401 | 72 | 57.992 | 0.805 | 216 | 79.304 | 0.367 | 0.73 |
| 8 | 397 | 171.5 | 0.432 | 207 | 178.561 | 0.863 | 240 | 67.782 | 0.282 | 2.63 |
| 9 | 251 | 111.47 | 0.444 | 122 | 95.650 | 0.784 | 171 | 71.104 | 0.416 | 1.35 |
| 10 | 374 | 156.63 | 0.419 | 145 | 131.186 | 0.905 | 265 | 106.481 | 0.402 | 1.23 |
| 11 | 218 | 116 | 0.532 | 123 | 92.466 | 0.752 | 125 | 76.28 | 0.610 | 1.21 |
| 12 | 274 | 172.04 | 0.628 | 144 | 164.174 | 1.140 | 174 | 96.972 | 0.557 | 1.69 |
| 13 | 247 | 129.08 | 0.523 | 135 | 119.459 | 0.885 | 151 | 64.097 | 0.424 | 1.86 |
| 14 | 377 | 156.6 | 0.415 | 183 | 158.806 | 0.868 | 230 | 64.863 | 0.282 | 2.45 |
| 15 | 324 | 132.01 | 0.407 | 160 | 125.678 | 0.785 | 208 | 76.551 | 0.368 | 1.64 |
| 16 | 339 | 164.2 | 0.484 | 183 | 157.621 | 0.861 | 210 | 90.283 | 0.430 | 1.75 |
| 17 | 247 | 158.4 | 0.641 | 144 | 153.737 | 1.068 | 151 | 82.783 | 0.548 | 1.86 |
| 18 | 218 | 105.59 | 0.484 | 109 | 98.316 | 0.902 | 143 | 77.2 | 0.540 | 1.27 |
| 19 | 183 | 120.61 | 0.659 | 82 | 70.275 | 0.857 | 124 | 86.103 | 0.694 | 0.82 |
| 20 | 99 | 78.95 | 0.797 | 82 | 81.608 | 0.995 | 44 | 43.736 | 0.994 | 1.87 |
| 21 | 136 | 145.33 | 1.069 | 136 | 151.049 | 1.111 | 36 | 40.997 | 1.139 | 3.68 |
| 22 | 328 | 207.76 | 0.633 | 162 | 197.049 | 1.216 | 204 | 82.058 | 0.402 | 2.40 |
| Total | 6144 | 3181.54 | 0.518 | 3120 | 2865.965 | 0.919 | 3814 | 1746.597 | 0.458 | 1.64 |
| LG 1 | Consensus Map | Female Map | Male Map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <5 cM 2 | 5–10 cM | 10–20 cM | >20 cM | Ratio (%) 3 | <5 cM | 5–10 cM | 10–20 cM | >20 cM | Ratio (%) | <5 cM | 5–10 cM | 10–20 cM | >20 cM | Ratio (%) | |
| 1 | 516 | 5 | 1 | 0 | 98.9 | 297 | 11 | 3 | 0 | 95.5 | 264 | 4 | 0 | 0 | 98.5 |
| 2 | 275 | 5 | 1 | 0 | 97.9 | 130 | 5 | 2 | 0 | 94.9 | 184 | 3 | 0 | 0 | 98.4 |
| 3 | 398 | 1 | 0 | 0 | 99.7 | 186 | 3 | 0 | 0 | 98.4 | 226 | 3 | 0 | 0 | 98.7 |
| 4 | 333 | 4 | 1 | 0 | 98.5 | 172 | 4 | 2 | 0 | 96.6 | 199 | 2 | 0 | 0 | 99.0 |
| 5 | 189 | 3 | 1 | 0 | 97.9 | 85 | 6 | 1 | 0 | 92.4 | 112 | 2 | 0 | 0 | 98.3 |
| 6 | 130 | 2 | 1 | 0 | 97.7 | 17 | 2 | 0 | 0 | 89.5 | 114 | 2 | 2 | 0 | 96.6 |
| 7 | 259 | 1 | 0 | 0 | 99.6 | 71 | 0 | 0 | 0 | 100.0 | 213 | 2 | 0 | 0 | 99.1 |
| 8 | 390 | 4 | 2 | 0 | 98.5 | 197 | 5 | 4 | 0 | 95.6 | 238 | 0 | 1 | 0 | 99.6 |
| 9 | 246 | 3 | 1 | 0 | 98.4 | 115 | 5 | 1 | 0 | 95.0 | 167 | 3 | 0 | 0 | 98.2 |
| 10 | 369 | 3 | 1 | 0 | 98.9 | 137 | 6 | 1 | 0 | 95.1 | 261 | 0 | 3 | 0 | 98.9 |
| 11 | 215 | 2 | 0 | 0 | 99.1 | 118 | 4 | 0 | 0 | 96.7 | 122 | 2 | 0 | 0 | 98.4 |
| 12 | 266 | 7 | 0 | 0 | 97.4 | 133 | 10 | 0 | 0 | 93.0 | 169 | 3 | 1 | 0 | 97.7 |
| 13 | 244 | 2 | 0 | 0 | 99.2 | 131 | 2 | 1 | 0 | 97.8 | 149 | 1 | 0 | 0 | 99.3 |
| 14 | 374 | 2 | 0 | 0 | 99.5 | 178 | 4 | 0 | 0 | 97.8 | 228 | 0 | 1 | 0 | 99.6 |
| 15 | 320 | 3 | 0 | 0 | 99.1 | 153 | 4 | 2 | 0 | 96.2 | 206 | 1 | 0 | 0 | 99.5 |
| 16 | 335 | 3 | 0 | 0 | 99.1 | 177 | 4 | 1 | 0 | 97.3 | 206 | 3 | 0 | 0 | 98.6 |
| 17 | 241 | 5 | 0 | 0 | 98.0 | 137 | 5 | 0 | 1 | 95.8 | 147 | 3 | 0 | 0 | 98.0 |
| 18 | 215 | 2 | 0 | 0 | 99.1 | 106 | 2 | 0 | 0 | 98.2 | 141 | 1 | 0 | 0 | 99.3 |
| 19 | 176 | 3 | 3 | 0 | 96.7 | 78 | 2 | 1 | 0 | 96.3 | 117 | 4 | 2 | 0 | 95.1 |
| 20 | 97 | 0 | 1 | 0 | 99.0 | 77 | 3 | 1 | 0 | 95.1 | 41 | 1 | 1 | 0 | 95.4 |
| 21 | 129 | 6 | 0 | 0 | 95.6 | 129 | 6 | 0 | 0 | 95.6 | 32 | 3 | 0 | 0 | 91.4 |
| 22 | 322 | 5 | 0 | 0 | 98.5 | 154 | 6 | 1 | 0 | 95.7 | 202 | 0 | 1 | 0 | 99.5 |
| Total | 6039 | 71 | 13 | 0 | 98.5 | 2978 | 99 | 21 | 1 | 95.8 | 3738 | 43 | 12 | 0 | 98.0 |
| Trait | LG | Position | Marker | LOD 1 | Variance | PVE (%) 2 | Left Marker | Right Marker |
|---|---|---|---|---|---|---|---|---|
| Body weight | LG3 | 30.36 | lm2808 | 3.13 | 2.54 | 10.6 | lm2619 | lm2621 |
| LG5 | 4.44 | lm2694 | 3.19 | 2.54 | 10.8 | lm3003 | lm665 | |
| LG6 | 52.98 | lm829 | 3.79 | 2.48 | 12.6 | lm1320 | lm1656 | |
| LG7 | 80.07 | lm3265 | 3.1 | 2.54 | 10.5 | lm2280 | hk690 | |
| LG7 | 90.37 | np2126 | 3.33 | 2.52 | 11.2 | np1854 | np1510 | |
| LG7 | 94.69 | lm575 | 3.28 | 2.53 | 11 | lm541 | lm574 | |
| LG18 | 47.37 | hk521 | 3.15 | 2.54 | 10.6 | lm2106 | lm2035 | |
| LG18 | 65.19 | lm864 | 5.06 | 2.37 | 16.5 | lm863 | np586 | |
| LG18 | 70.79 | lm1135 | 3.9 | 2.47 | 13 | np784 | hk284 | |
| LG22 | 198.7 | np845 | 3.11 | 2.54 | 10.5 | np1706 | np837 | |
| Body length | LG5 | 1.6 | lm2222 | 4.47 | 1.24 | 14.8 | lm3000 | lm3002 |
| LG5 | 4.44 | lm2694 | 4.34 | 1.25 | 14.4 | lm3003 | lm665 | |
| LG5 | 15.68 | lm1384 | 3.73 | 1.28 | 12.6 | hk711 | lm1569 | |
| LG18 | 65.08 | lm863 | 3.5 | 1.29 | 11.8 | hk520 | lm864 | |
| LG20 | 28.98 | np2401 | 3.05 | 1.31 | 10.4 | np787 | np1754 | |
| LG22 | 198.7 | np845 | 3.06 | 1.31 | 10.4 | np1706 | np837 | |
| Papillae number | LG5 | 4.44 | lm2694 | 3.57 | 43.36 | 12.1 | lm3003 | lm665 |
| LG6 | 53.03 | lm832 | 3.95 | 42.78 | 13.2 | lm1656 | lm1834 | |
| LG11 | 93.65 | lm2134 | 3.85 | 42.93 | 12.9 | lm1742 | hk196 | |
| LG18 | 53.03 | lm2110 | 3.17 | 44 | 10.8 | lm2111 | lm2109 | |
| LG18 | 65.08 | lm863 | 4.13 | 42.5 | 13.8 | hk520 | lm864 | |
| LG22 | 198.7 | np845 | 3.52 | 43.45 | 11.9 | np1706 | np837 |
| Marker | LG | Scaffold | Annotation | GO | KEGG | ||
|---|---|---|---|---|---|---|---|
| Cellular Component | Biological Process | Molecular Function | |||||
| lm2808 | lg3 | 1093 | - | - | - | - | - |
| lm2694 | lg5 | 996 | CREB3L1 | transcription; transcription regulation; unfolded protein response; | activator; developmental protein; DNA-binding; | cAMP signaling pathway; growth hormone synthesis, secretion and action; | |
| lm829 | lg6 | 153 | caveolin-1 | endoplasmic reticulum membrane; Golgi membrane; caveolar macromolecular signaling complex; endocytic vesicle membrane; | apoptotic signaling pathway; cell differentiation; inactivation of MAPK activity; negative regulation of BMP and Wnt signaling pathway; | ATPase binding; signaling receptor binding; cell differentiation; regulation of canonical Wnt signaling pathway; molecular adaptor activity; | endocytosis; focal adhesion; transport and catabolism; genetic information processing; |
| lm3265 | lg7 | 1691 | - | - | - | - | - |
| np2126 | lg7 | 1232 | - | - | - | - | - |
| lm575 | lg7 | 87 | Transposon-derived Buster3 transposase-like protein | nucleoplasm; | protein binding; | signaling and cellular processes; | |
| hk521 | lg18 | 562 | homogentisate 1,2-dioxygenase | cytosol; extracellular exosome; cytoplasm; | L-phenylalanine catabolic process; tyrosine catabolic process; | identical protein binding; metal ion binding; | alkaptonuria; tyrosine metabolism; |
| lm864 | lg18 | 157 | protein still life, isoforms C/SIF type 2 | membrane; synapse; | actin cytoskeleton organization; signal transduction; cell junction; synapse formation | guanyl-nucleotide exchange factor activity; | signaling and cellular processes; Dbl-Like RhoGEF family proteins; |
| lm1135 | lg18 | 233 | - | - | |||
| np845 | lg22 | 252 | transcription factor 4 isoform X3 | ||||
| lm2222 | lg5 | 615 | nerve growth factor-like protein | axon; dendrite; synaptic vesicle; | modulation of chemical synaptic transmission; nerve development; nerve growth factor signaling pathway; | growth factor activity; nerve growth factor receptor binding; | MAPK signaling pathway; calcium signaling pathway; cytokines and growth factors; intramolecular chaperones; apoptosis; |
| lm2694 | lg5 | 996 | CREB3L1 | ||||
| lm1384 | lg5 | 300 | IgGFc-binding protein | extracellular matrix; extracellular exosome; extracellular space; | |||
| lm863 | lg18 | 157 | protein still life, isoforms C/SIF type 2 | ||||
| np2401 | lg20 | 1900 | nucleoside diphosphate kinase 7 | mitochondrial inner membrane; mitochondrion; | lipid transport; nucleotide metabolism; transport; transcription regulation; | ATP binding; metal ion binding; | purine metabolism; membrane trafficking; |
| np845 | lg22 | 252 | transcription factor 4 isoform X3 | ||||
| lm2694 | lg5 | 996 | CREB3L1 | ||||
| lm832 | lg6 | 153 | hyalin-like | integral component ofmembrane; | |||
| lm2134 | lg11 | 576 | neural cell adhesion molecule 1-like | cell membrane; | axonal fasciculation; cell adhesion; neurogenesis; | identical protein binding; | cell adhesion molecules; |
| lm2110 | lg18 | 562 | - | - | - | - | - |
| lm863 | lg18 | 157 | protein still life, isoforms C/SIF type 2 | ||||
| np845 | lg22 | 252 | transcription factor 4 isoform X3 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, W.; Huo, D.; Liu, S.; Xing, L.; Su, F.; Yang, H.; Sun, L. Construction of a High-Density Genetic Linkage Map for the Mapping of QTL Associated with Growth-Related Traits in Sea Cucumber (Apostichopus japonicus). Biology 2022, 11, 50. https://doi.org/10.3390/biology11010050
Cui W, Huo D, Liu S, Xing L, Su F, Yang H, Sun L. Construction of a High-Density Genetic Linkage Map for the Mapping of QTL Associated with Growth-Related Traits in Sea Cucumber (Apostichopus japonicus). Biology. 2022; 11(1):50. https://doi.org/10.3390/biology11010050
Chicago/Turabian StyleCui, Wei, Da Huo, Shilin Liu, Lili Xing, Fang Su, Hongsheng Yang, and Lina Sun. 2022. "Construction of a High-Density Genetic Linkage Map for the Mapping of QTL Associated with Growth-Related Traits in Sea Cucumber (Apostichopus japonicus)" Biology 11, no. 1: 50. https://doi.org/10.3390/biology11010050