
Comparative Assessment of NMR Probes for the Experimental Description of Protein Folding Pathways with High-Pressure NMR

 ,
,  and
and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Protein NMR Resonance Assignment
2.3. Protein Unfolding
3. Results
3.1. NMR Resonance Assignment
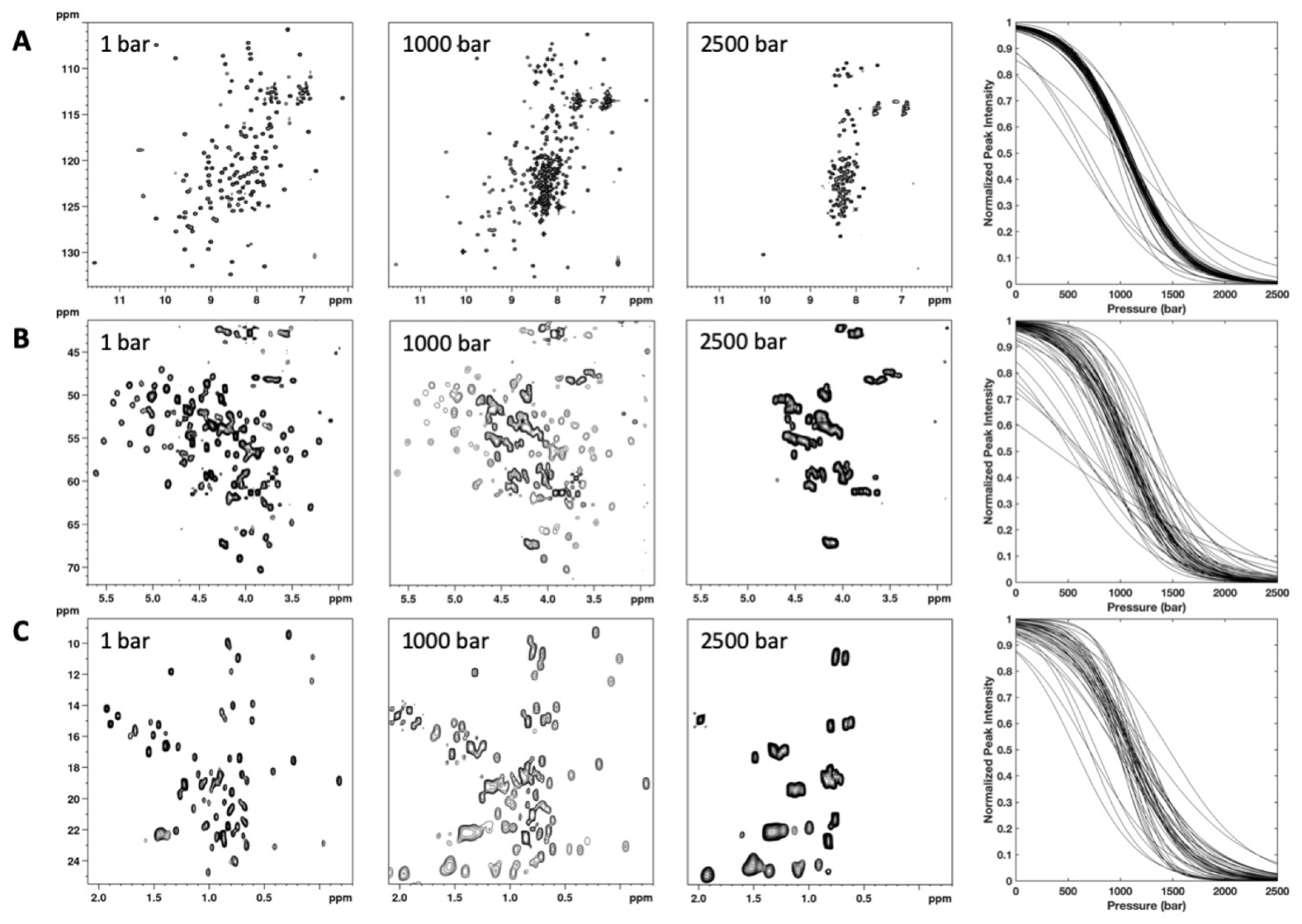
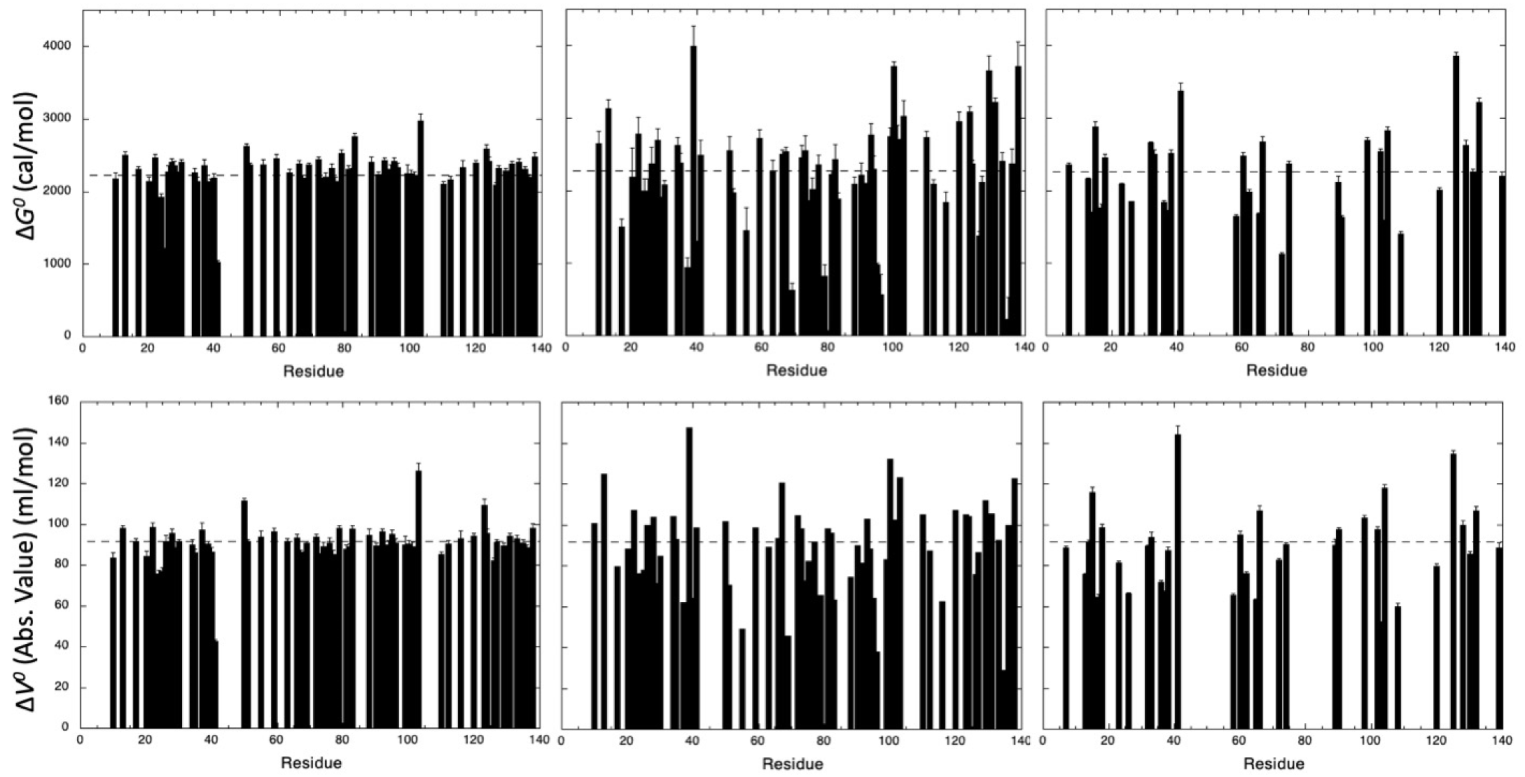
3.2. HP-NMR Denaturation Study: Measuring the Thermodynamic Parameters for the Unfolding Reaction
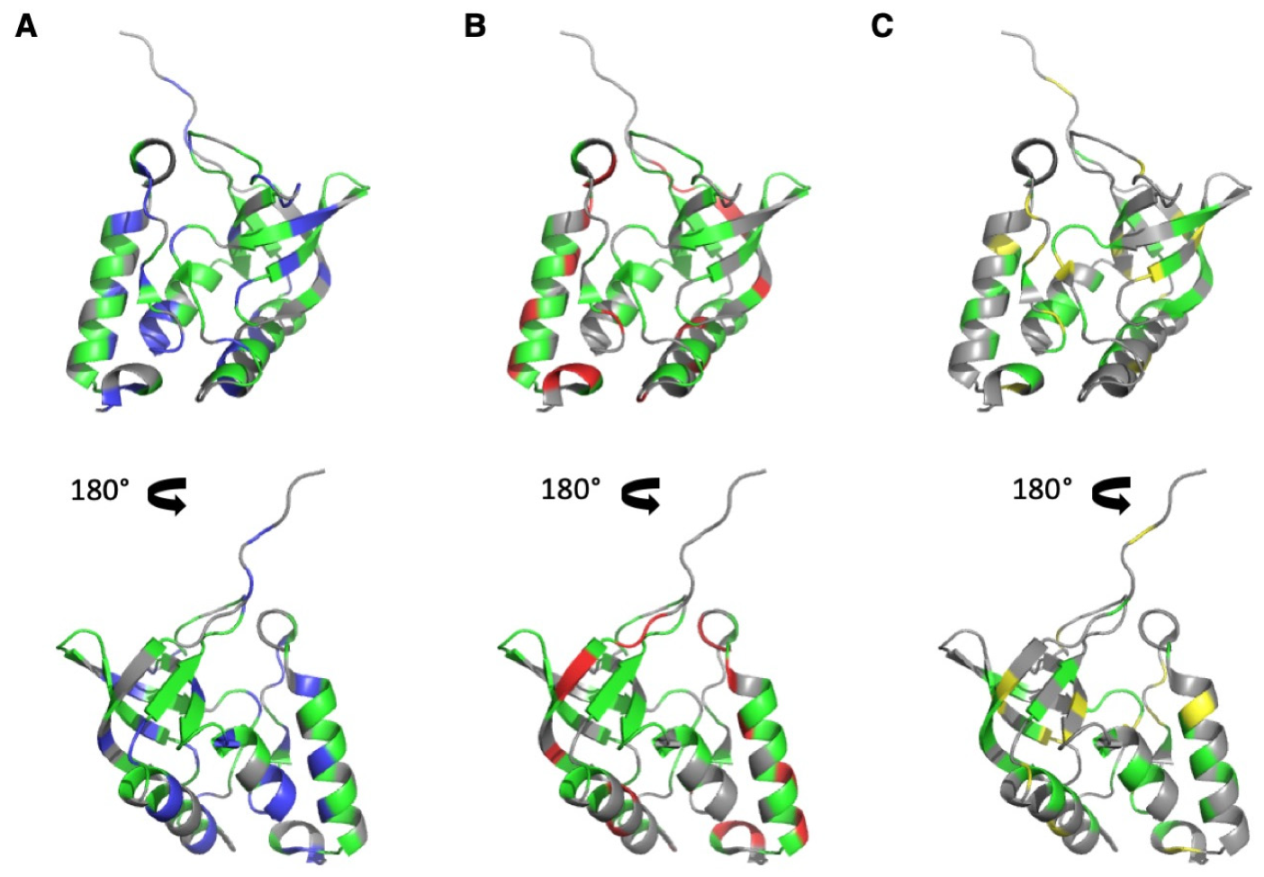
3.3. HP-NMR Denaturation Study: Exploring the Folding Pathways of ∆+PHS SNase
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heremans, K.; Smeller, L. Protein structure and dynamics at high pressure. Biochim. Biophys. Acta 1998, 1386, 353–370. [Google Scholar] [CrossRef]
- Kamatari, Y.O.; Kitahara, R.; Yamada, H.; Yokoyama, S.; Akasaka, K. High-pressure NMR spectroscopy for characterizing folding intermediates and denatured states of proteins. Methods 2004, 34, 133–143. [Google Scholar] [CrossRef]
- Balny, C.; Masson, P.; Heremans, K. High pressure effects on biological macromolecules: From structural changes to alteration of cellular processes. Biochim. Biophys. Acta 2002, 1595, 3–10. [Google Scholar] [CrossRef]
- Akasaka, K.; Kitahara, R.; Kamatari, Y.O. Exploring the folding energy landscape with pressure. Arch. Biochem. Biophys. 2013, 531, 110–115. [Google Scholar] [CrossRef]
- Royer, C.A. Revisiting volume changes in pressure-induced protein unfolding. Biochim. Biophys. Acta 2002, 1595, 201–209. [Google Scholar] [CrossRef]
- Meersman, F.; Dobson, C.M.; Heremans, K. Protein unfolding, amyloid fibril formation and configurational energy landscapes under high pressure conditions. Chem. Soc. Rev. 2006, 35, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Royer, C.A. Why and How Does Pressure Unfold Proteins? Subcell. Biochem. 2015, 72, 59–71. [Google Scholar]
- Rouget, J.; Aksel, T.; Roche, J.; Saldana, J.; Garcia, A.E.; Barrick, D.; Royer, C.A. Size and sequence and the volume change of protein folding. J. Am. Chem. Soc. 2011, 133, 6020–6027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, J.; Dellarole, M.J.; Caro, A.; Guca, E.; Norberto, D.R.; Yang, Y.-S.; Garcia, A.E.; Roumestand, C.; García-Moreno, B.; Royer, C.A. Remodeling of the folding free-energy landscape of staphylococcal nuclease by cavity-creating mutations. Biochemistry 2012, 51, 9535–9546. [Google Scholar] [CrossRef]
- Roche, J.; Caro, J.A.; Norberto, D.R.; Barthe, P.; Roumestand, C.; Schlessman, J.L.; Garcia, A.E.; García-Moreno, B.E.; Royer, C.A. Cavities determine the pressure unfolding of proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 6945–6950. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Wakamoto, T.; Kejlberg, C.; Yoshimura, Y.; Nielsen, T.A.; Risor, M.W.; Sanggaard, K.W.; Kitahara, R.; Mulder, F.A.A. How internal cavities destabilize a protein. Proc. Natl. Acad. Sci. USA 2019, 116, 21031–21036. [Google Scholar] [CrossRef] [Green Version]
- Jonas, J.; Jonas, A. High-pressure NMR spectroscopy of proteins and membranes. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 287–318. [Google Scholar] [CrossRef]
- Akasaka, K.; Yamada, H. On-line cell high-pressure nuclear magnetic resonance technique: Application to protein studies. Methods Enzymol. 2001, 338, 134–158. [Google Scholar]
- Kremer, W. High-pressure NMR studies in proteins. Annu. Rep. NMR Spectrosc. 2006, 57, 177–203. [Google Scholar]
- Roche, J.; Royer, C.A.; Roumestand, C. Monitoring protein folding through high pressure NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 102–103, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Caro, J.A.; Wand, A.J. Practical aspects of high-pressure NMR spectroscopy and its applications in protein biophysics and structural biology. Methods 2018, 148, 67–80. [Google Scholar] [CrossRef]
- Roche, J.; Royer, C.A.; Roumestand, C. Exploring Protein Conformational Landscapes Using High-Pressure NMR. Methods Enzymol. 2019, 614, 293–320. [Google Scholar]
- Dubois, C.; Herrada, I.; Barthe, P.; Roumestand, C. Combining High-Pressure Perturbation with NMR Spectroscopy for a Structural and Dynamical Characterization of Protein Folding Pathways. Molecules 2020, 25, 5551. [Google Scholar] [CrossRef]
- Murzin, A.G. OB (oligonucleotide/olisaccharide binding)-fold: Common structural and functional solution for non-homologous sequences. EMBO J. 1993, 12, 861–867. [Google Scholar] [CrossRef]
- Arcus, V. OB-fold domains: A snapshot of the evolution of sequence, structure and function. Curr. Opin. Struct. Biol. 2002, 12, 794–801. [Google Scholar] [CrossRef]
- Taniuchi, H.; Anfinsen, C.B. An experimental approach to the study of the folding of the Staphylococcal nuclease. J. Biol. Chem. 1969, 244, 3864–3875. [Google Scholar] [CrossRef]
- Shortle, D.; Meeker, A.K. Residual structure in large fragments of staphylococcal nuclease: Effects of amino acids substitutions. Biochemistry 1998, 28, 936–944. [Google Scholar] [CrossRef]
- Alexandrescu, A.T.; Gittis, A.G.; Abeygunwardana, C.; Shortle, D. NMR structure of a stable OB-fold sub-domain isolated from staphylococcal nuclease. J. Mol. Biol. 1995, 250, 134–143. [Google Scholar] [CrossRef]
- Ye, K.; Jing, G.; Wang, J. Interactions between subdomains in the partially folded state of staphylococcal nuclease. Biochim. Biophys. Acta. 2000, 1479, 123–134. [Google Scholar] [CrossRef]
- Watson, E.; Matousek, W.M.; Irimies, E.L.; Alexandrescu, A.T. Partially folded states of staphylococcal nuclease highlight the conserved structural hierarchy of OB-fold proteins. Biochemistry 2007, 46, 9484–9494. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.-Y.; Wu, M.-C.; Fang, H.-J.; Forrest, M.D.; Hu, C.-K.; Tsong, T.Y.; Chen, H.M. The role of tryptophan in staphylococcal nuclease stability. Biophys. Chem. 2010, 151, 170–177. [Google Scholar] [CrossRef]
- Wang, M.; Feng, Y.; Yao, H.; Wang, J. Importance of the C-terminal loop L137-S141 for the folding and folding stability of staphylococcal nuclease stability. Biochemistry 2010, 49, 4318–4326. [Google Scholar] [CrossRef]
- Bedard, S.; Mayne, L.C.; Petersen, R.W.; Wand, A.J.; Englander, S.W. The foldon substructure of staphylococcal nuclease. J. Mol. Biol. 2008, 376, 1142–1154. [Google Scholar] [CrossRef] [Green Version]
- Roche, J.; Caro, J.A.; Dellarole, M.; Guca, E.; Royer, C.A.; García-Moreno, B.; Garcia, A.E. Roumestand, C. Structural, energetic and dynamic responses of the native state ensemble of staphylococcal nuclease to cavity-creating mutations. Proteins 2013, 81, 1069–1080. [Google Scholar] [CrossRef]
- Shortle, D.; Meeker, A.K.; Freire, E. Stability mutants of staphylococcal nuclease: Large compensating enthalpy-entropy changes for the reversible denaturation reaction. Biochemistry 1988, 27, 4761–4768. [Google Scholar] [CrossRef]
- Pons, J.L.; Malliavin, T.E.; Delsuc, M.A. Gifa V. 4: A complete package for NMR data set processing. J. Biomol. NMR 1996, 8, 445–452. [Google Scholar] [CrossRef]
- Fossat, M.J.; Dao, T.P.; Jenkins, K.; Dellarole, M.; Yang, Y.-S.; McCallum, S.A.; Garcia, A.E.; Barrick, D.; Roumestand, C.; Royer, C.A. High-Resolution Mapping of a Repeat Protein Folding Free Energy Landscape. Biophys. J. 2016, 111, 2368–2376. [Google Scholar] [CrossRef]
- Jenkins, K.A.; Fossat, M.J.; Zhang, S.; Rai, D.K.; Klein, S.; Gillilan, R.; White, Z.; Gerlich, G.; McCallum, S.A.; Winter, R.; et al. The consequences of cavity creation on the folding landscape of a repeat protein depend upon context. Proc. Natl. Acad. Sci. USA. 2018, 115, 8153–8161. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, E.J.; Wand, A.J. Local stability and dynamics of apocytochrome b562 examined by the dependence of hydrogen exchange on hydrostatic pressure. Biochemistry 1998, 37, 9877–9883. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Deuren, V.; Yang, Y.-S.; de Guillen, K.; Dubois, C.; Royer, C.A.; Roumestand, C.; Barthe, P. Comparative Assessment of NMR Probes for the Experimental Description of Protein Folding Pathways with High-Pressure NMR. Biology 2021, 10, 656. https://doi.org/10.3390/biology10070656
Van Deuren V, Yang Y-S, de Guillen K, Dubois C, Royer CA, Roumestand C, Barthe P. Comparative Assessment of NMR Probes for the Experimental Description of Protein Folding Pathways with High-Pressure NMR. Biology. 2021; 10(7):656. https://doi.org/10.3390/biology10070656
Chicago/Turabian StyleVan Deuren, Vincent, Yin-Shan Yang, Karine de Guillen, Cécile Dubois, Catherine Anne Royer, Christian Roumestand, and Philippe Barthe. 2021. "Comparative Assessment of NMR Probes for the Experimental Description of Protein Folding Pathways with High-Pressure NMR" Biology 10, no. 7: 656. https://doi.org/10.3390/biology10070656