Secondary Structure of the Novel Myosin Binding Domain WYR and Implications within Myosin Structure


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Purification of MHC Fragment
2.2. WYR Peptide
2.3. Circular Dichroism Sample Preparation
2.4. Circular Dichroism Measurements and Analysis
2.5. Separated Parameters Method
2.6. Combined Parameters Method
2.7. Co-sedimentation Assays
2.8. Densitometry
3. Results
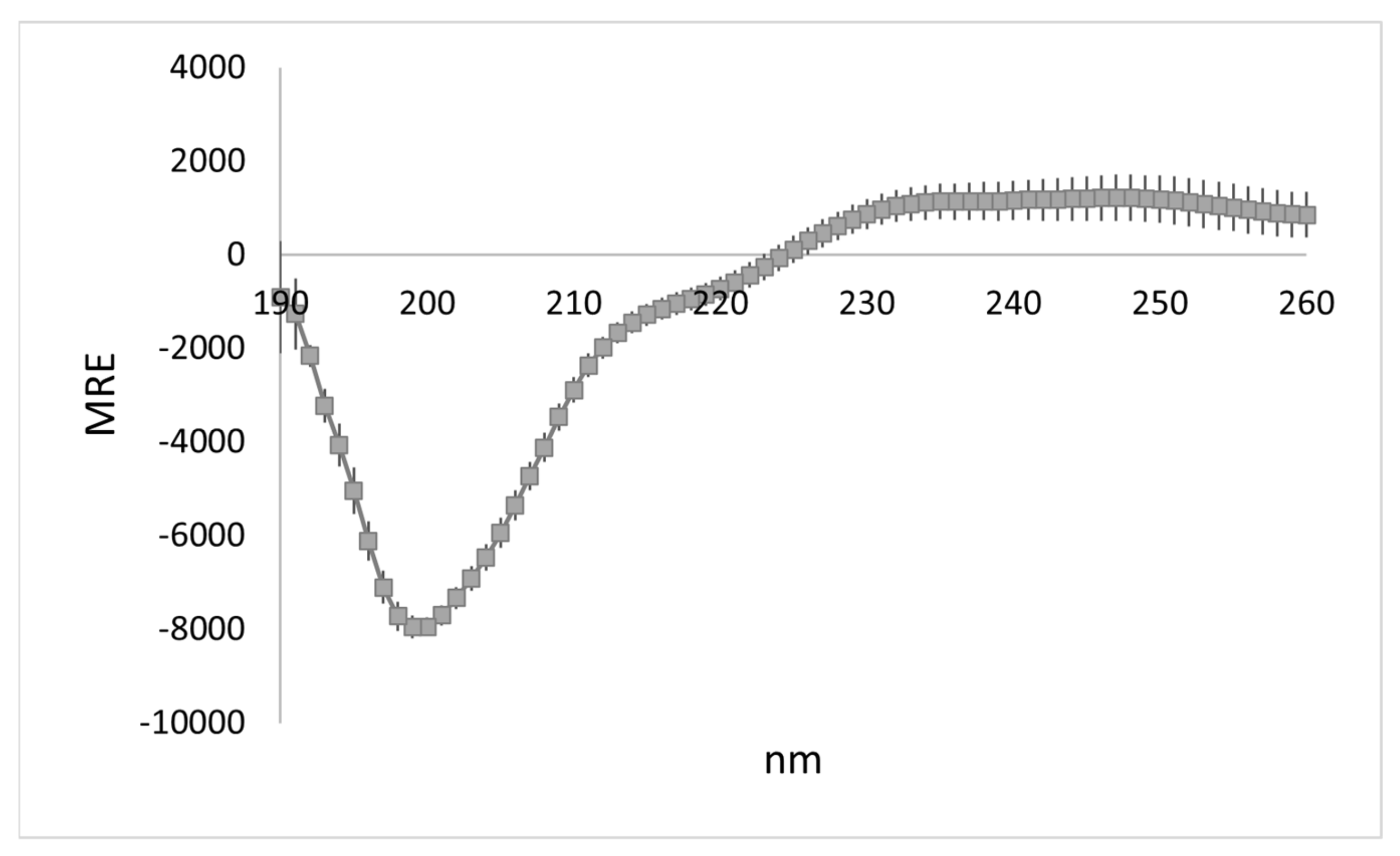
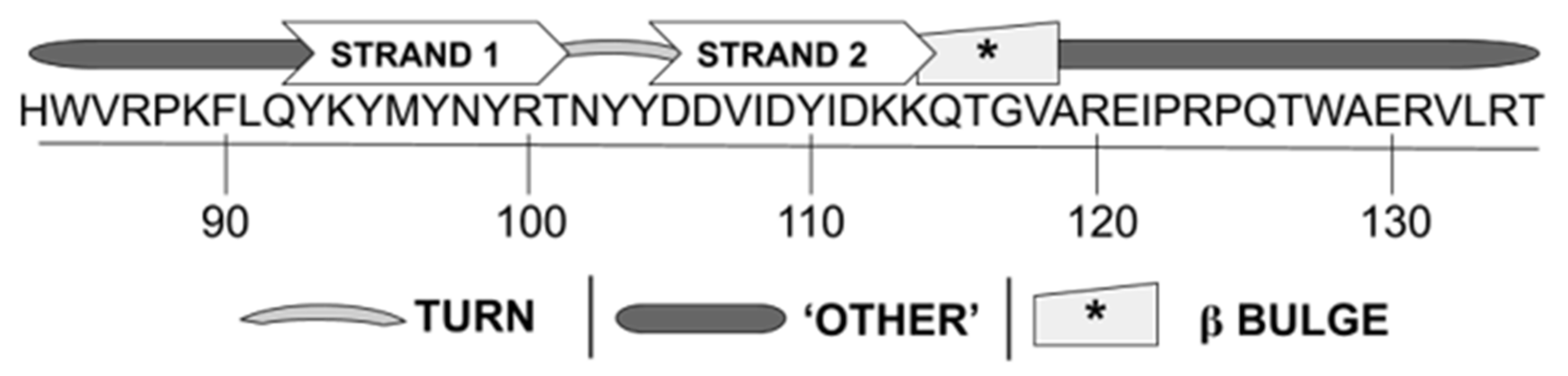
3.1. WYR Structure Contains Antiparallel Beta Strands and Aperiodic Elements
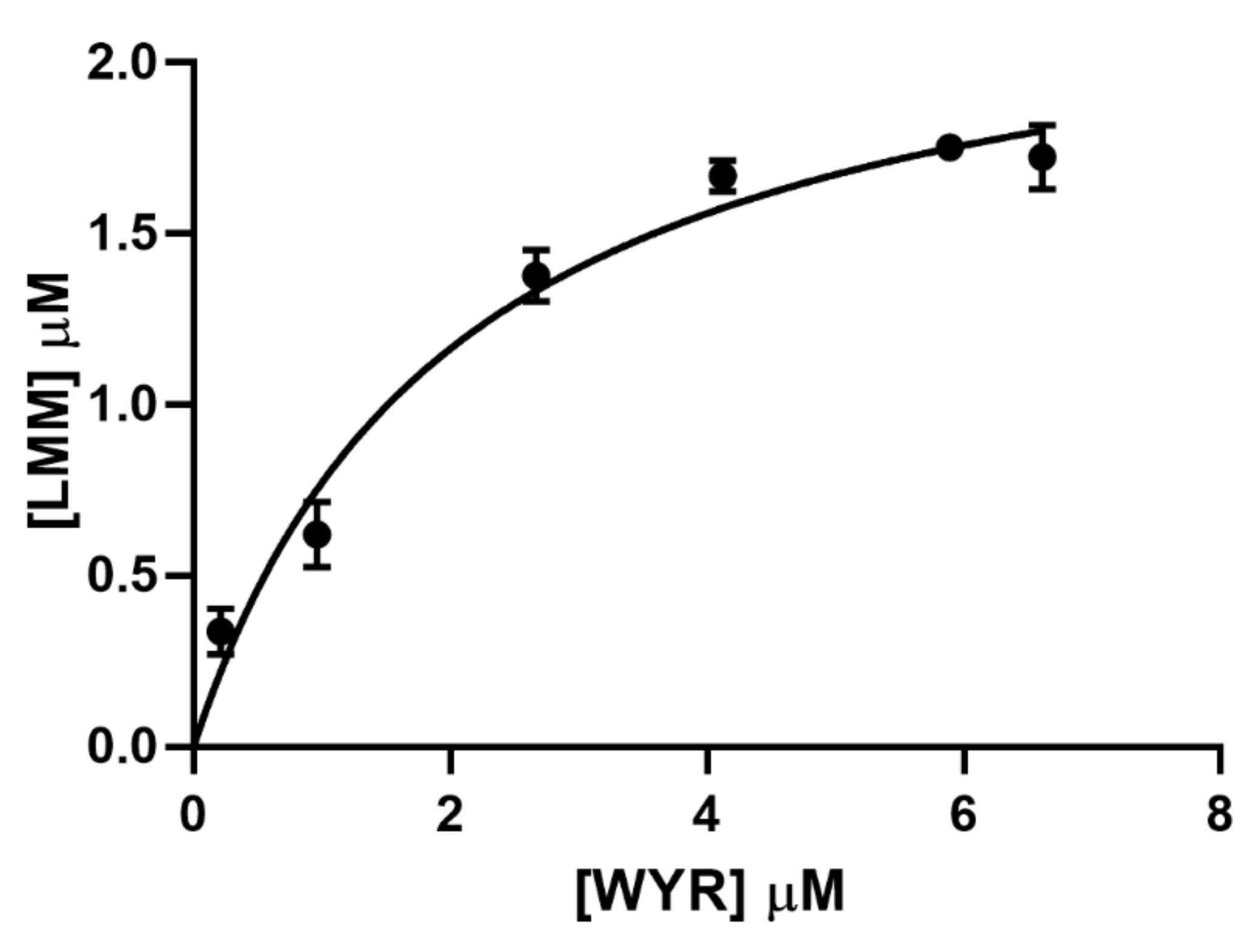
3.2. WYR Binds the LMM
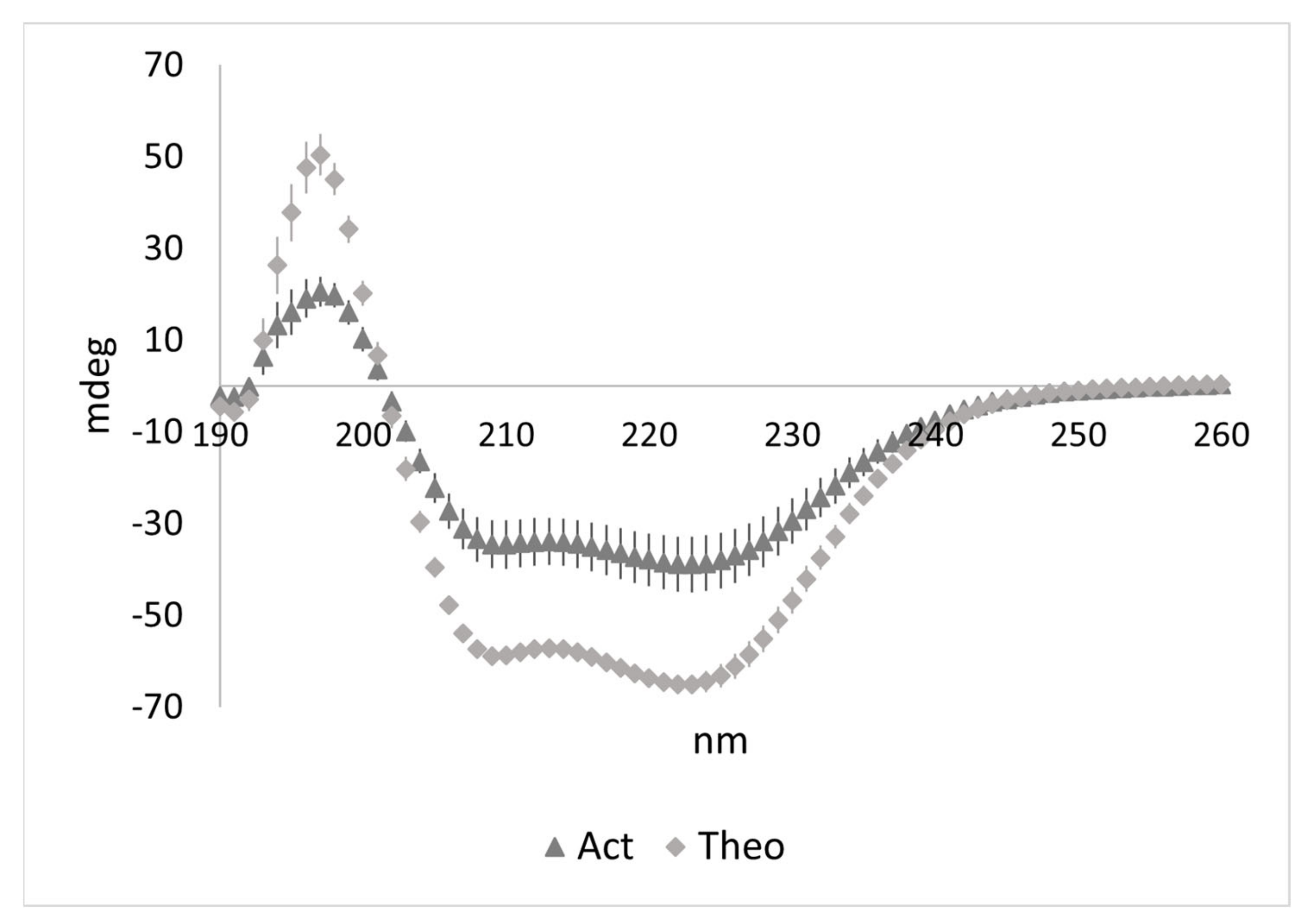
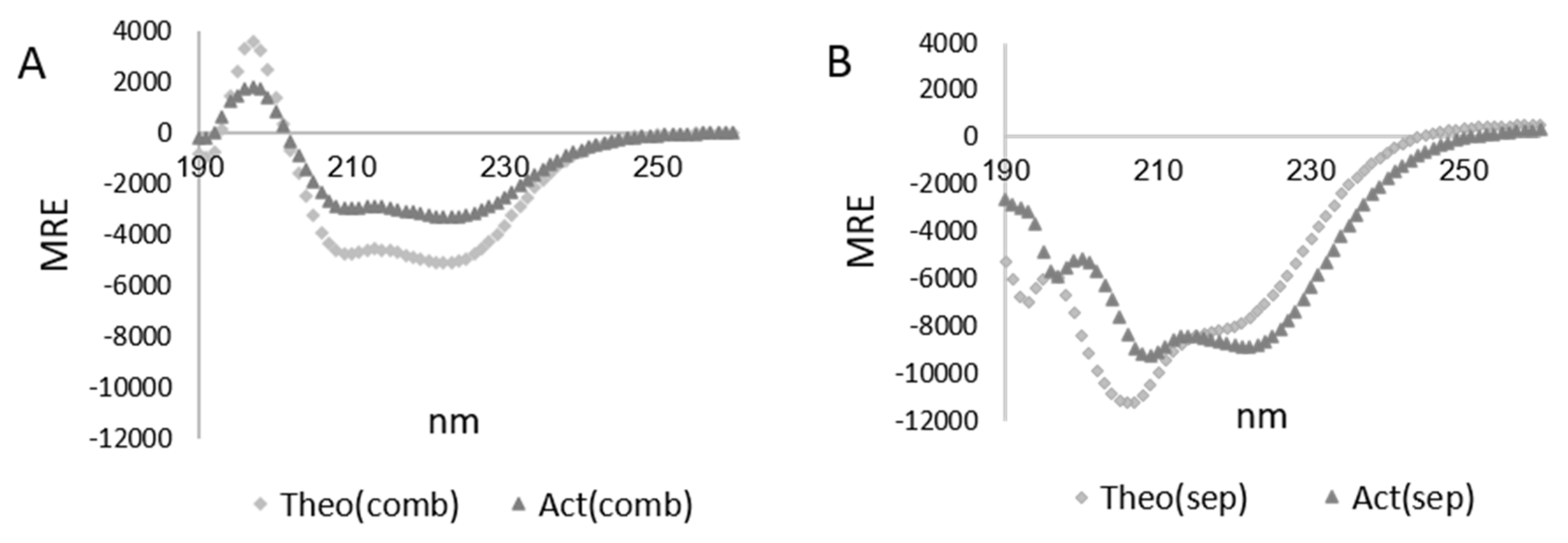
3.3. Characterization of the LMMWYR Structure
3.4. Characterization of LMM Structure
4. Discussion
4.1. WYR Structure
4.2. LMMWYR Structure
4.3. Implications for Thick Filament Structure and Mechanics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iwamoto, H. Structure, function and evolution of insect flight muscle. Biophysics 2011, 7, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Sotavalta, O. Recordings of high wing-stroke and thoracic vibration frequency in some midges. Biol. Bull. 1953, 104, 439–444. [Google Scholar] [CrossRef]
- Kreipke, R.; Kwon, Y.; Shcherbata, H.; Ruohola-Baker, H. Drosophila Melanogaster as a Model of Muscle Degeneration Disorders; Elsevier BV: New York, NY, USA, 2017; Volume 121, pp. 83–109. [Google Scholar]
- A Kronert, W.; Bell, K.M.; Viswanathan, M.C.; Melkani, G.C.; Trujillo, A.S.; Huang, A.; Melkani, A.; Cammarato, A.; Swank, D.M.; I Bernstein, S. Prolonged cross-bridge binding triggers muscle dysfunction in a Drosophila model of myosin-based hypertrophic cardiomyopathy. eLife 2018, 7, 7. [Google Scholar] [CrossRef]
- Mohr, S.E. First in Fly: Drosophila Research and Biological Discovery; Harvard University Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Sink, H. Muscle Development in Drosophila. In Muscle Development in Drosophila; Springer Science and Business Media LLC: New York, NY, USA, 2006. [Google Scholar]
- Vigoreaux, J. Nature’s Versatile Engine: Insect Flight Muscle Inside and Out; Springer Science & Business Media, Inc.: New York, NY, USA, 2007; Volume 288. [Google Scholar]
- Ayer, G.; Vigoreaux, J.O. Flightin is a Myosin Rod Binding Protein. Cell Biophys. 2003, 38, 41–54. [Google Scholar] [CrossRef]
- Reedy, M.C.; Bullard, B.; Vigoreaux, J.O. Flightin Is Essential for Thick Filament Assembly and Sarcomere Stability in Drosophila Flight Muscles. J. Cell Biol. 2000, 151, 1483–1500. [Google Scholar] [CrossRef]
- Soto-Adames, F.N.; Alvarez-Ortiz, P.; Vigoreaux, J.O. An Evolutionary Analysis of Flightin Reveals a Conserved Motif Unique and Widespread in Pancrustacea. J. Mol. Evol. 2013, 78, 24–37. [Google Scholar] [CrossRef]
- Vigoreaux, J.O.; Hernandez, C.; Moore, J.; Ayer, G.; Maughan, D. A genetic deficiency that spans the flightin gene of Drosophila melanogaster affects the ultrastructure and function of the flight muscles. J. Exp. Biol. 1998, 201, 2033–2044. [Google Scholar] [CrossRef]
- Chakravorty, S.; Tanner, B.; Foelber, V.L.; Vu, H.; Rosenthal, M.; Ruiz, T.; Vigoreaux, J.O. Flightin maintains myofilament lattice organization required for optimal flight power and courtship song quality in Drosophila. Proc. R. Soc. Boil. Sci. 2017, 284, 20170431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, D.G.M.S. Evolution of the Insects, 1st ed.; Cambridge University Press: New York, NY, USA, 2005. [Google Scholar]
- Henkin, J.A.; Maughan, D.W.; Vigoreaux, J.O. Mutations that affect flightin expression in Drosophila alter the viscoelastic properties of flight muscle fibers. Am. J. Physiol. Cell Physiol. 2004, 286, C65–C72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, B.; Ayer, G.; Heymann, N.; Maughan, D.W.; Lehmann, F.-O.; Vigoreaux, J.O. Flight muscle properties and aerodynamic performance of Drosophila expressing a flightin transgene. J. Exp. Biol. 2005, 208, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, B.C.W.; Miller, M.S.; Miller, B.M.; Lekkas, P.; Irving, T.C.; Maughan, D.W.; Vigoreaux, J.O. COOH-terminal truncation of flightin decreases myofilament lattice organization, cross-bridge binding, and power output in Drosophila indirect flight muscle. Am. J. Physiol. Physiol. 2011, 301, C383–C391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasek, N.S.; Nyland, L.R.; Vigoreaux, J.O. The Contributions of the Amino and Carboxy Terminal Domains of Flightin to the Biomechanical Properties of Drosophila Flight Muscle Thick Filaments. Biology 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contompasis, J.L.; Nyland, L.R.; Maughan, D.W.; Vigoreaux, J.O. Flightin Is Necessary for Length Determination, Structural Integrity, and Large Bending Stiffness of Insect Flight Muscle Thick Filaments. J. Mol. Biol. 2010, 395, 340–348. [Google Scholar] [CrossRef]
- Kronert, W.A.; O’Donnell, P.T.; Fieck, A.; Lawn, A.; Vigoreaux, J.O.; Sparrow, J.C.; Bernstein, S.I. Defects in theDrosophilaMyosin Rod Permit Sarcomere Assembly but Cause Flight Muscle Degeneration. J. Mol. Biol. 1995, 249, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Daneshparvar, N.; Taylor, D.W.; O’Leary, T.S.; Rahmani, H.; Abbasiyeganeh, F.; Previs, M.J.; Taylor, K.A.; Cryo, E.M. Structure of Drosophila Flight Muscle Thick Filaments at 7Å Resolution. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hu, Z.; Taylor, D.W.; Reedy, M.K.; Edwards, R.J.; Taylor, K.A. Structure of myosin filaments from relaxed Lethocerus flight muscle by cryo-EM at 6 Å resolution. Sci. Adv. 2016, 2, e1600058. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, E.N.; Taylor, K.C.; Andreas, M.P.; Ajay, G.; Heinze, N.T.; Cui, Q.; Rayment, I. A composite approach towards a complete model of the myosin rod. Proteins Struct. Funct. Bioinform. 2015, 84, 172–189. [Google Scholar] [CrossRef] [Green Version]
- Briggs, M. Densitometry using ImageJ. Available online: https://www.sybil-fp7.eu/node/95 (accessed on 1 April 2019).
- Starna Cells, I. Suggestions for Cleaning Cells. Available online: http://www.starnacells.com/d_tech/tech01.html (accessed on 25 September 2017).
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [Green Version]
- Wolny, M.; Colegrave, M.; Colman, L.; White, E.; Knight, P.J.; Peckham, M. Cardiomyopathy mutations in the tail of beta-cardiac myosin modify the coiled-coil structure and affect integration into thick filaments in muscle sarcomeres in adult cardiomyocytes. J. Biol. Chem. 2013, 288, 31862–31952. [Google Scholar] [CrossRef] [Green Version]
- Armel, T.Z.; Leinwand, L.A. Mutations in the beta-myosin rod cause myosin storage myopathy via multiple mechanisms. Proc. Natl. Acad. Sci. USA 2009, 106, 6291–6296. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Thyparambil, A.; Latour, R.A. Protein helical structure determination using CD spectroscopy for solutions with strong background absorbance from 190 to 230nm. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 2331–2337. [Google Scholar] [CrossRef] [Green Version]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2007, 89, 392–400. [Google Scholar] [CrossRef]
- Petersen, B.; Lundegaard, C.; Petersen, T.N. NetTurnP—Neural Network Prediction of Beta-turns by Use of Evolutionary Information and Predicted Protein Sequence Features. PLoS ONE 2010, 5, e15079. [Google Scholar] [CrossRef] [Green Version]
- Zondlo, N.J. Aromatic–Proline Interactions: Electronically Tunable CH/π Interactions. Accounts Chem. Res. 2013, 46, 1039–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashok Kumar, T. CFSSP: Chou and Fasman Secondary Structure Prediction server. WIDE SPECTRUM Res. J. 2013, 1, 15–19. [Google Scholar] [CrossRef]
- Lin, K.; Simossis, V.A.; Taylor, W.R.; Heringa, J. A simple and fast secondary structure prediction method using hidden neural networks. Bioinformatics 2004, 21, 152–159. [Google Scholar] [CrossRef] [Green Version]
- Rost, B.; Sander, C. Improved prediction of protein secondary structure by use of sequence profiles and neural networks. Proc. Natl. Acad. Sci. USA 1993, 90, 7558–7562. [Google Scholar] [CrossRef] [Green Version]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.; E Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuff, J.A.; Barton, G. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins Struct. Funct. Bioinform. 2000, 40, 502–511. [Google Scholar] [CrossRef]
- Cuff, J.A.; Clamp, M.E.; Siddiqui, A.S.; Finlay, M.; Barton, G. JPred: A consensus secondary structure prediction server. Bioinform. 1998, 14, 892–893. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deléage, G. NPS@: Network Protein Sequence Analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef]
- Yachdav, G.; Kloppmann, E.; Kajan, L.; Hecht, M.; Goldberg, T.; Hamp, T.; Hönigschmid, P.; Schafferhans, A.; Roos, M.; Bernhofer, M.; et al. PredictProtein—An open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014, 42, W337–W343. [Google Scholar] [CrossRef] [Green Version]
- Montgomerie, S.; Sundararaj, S.; Gallin, W.J.; Wishart, D.S. Improving the accuracy of protein secondary structure prediction using structural alignment. BMC Bioinform. 2006, 7, 301. [Google Scholar] [CrossRef] [Green Version]
- Newberry, R.W.; Raines, R.T. Secondary Forces in Protein Folding. ACS Chem. Biol. 2019, 14, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Looger, L.L.; Porter, L.L. Inaccurate secondary structure predictions often indicate protein fold switching. Protein Sci. 2019, 28, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Corrêa, D.; Ramos, C. The use of circular dichroism spectroscopy to study protein folding, form and function. Afr. J. Biochem. Res. 2009, 3, 164–173. [Google Scholar]
- Johnson, W.C. Analyzing protein circular dichroism spectra for accurate secondary structures. Proteins Struct. Funct. Bioinform. 1999, 35, 307–312. [Google Scholar] [CrossRef]
- Merelo, J.J.; Andrade, M.A.; Prieto, A.; Morán, F. Proteinotopic feature maps. Neurocomputing 1994, 6, 443–454. [Google Scholar] [CrossRef]
- Provencher, S.W.; Gloeckner, J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 1981, 20, 33–37. [Google Scholar] [CrossRef]
- Walters, J.; Milam, S.L.; Clark, A.C. Practical approaches to protein folding and assembly: Spectroscopic strategies in thermodynamics and kinetics. Methods Enzymol. 2009, 455, 1–39. [Google Scholar]
- Armel, T.Z.; Leinwand, L.A. A mutation in the beta-myosin rod associated with hypertrophic cardiomyopathy has an unexpected molecular phenotype. Biochem. Biophys. Res. Commun. 2010, 391, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, E.R.; Naik, A.R.; Lewis, B.; Kokotovich, K.M.; Li, M.; Stemmler, T.L.; Larsson, L.; Jena, B.P. Nanothermometry Reveals Calcium-Induced Remodeling of Myosin. Nano Lett. 2018, 18, 7021–7029. [Google Scholar] [CrossRef]
- Parker, F.; Batchelor, M.; Wolny, M.; Hughes, R.; Knight, P.J.; Peckham, M. A1603P and K1617del, Mutations in β-Cardiac Myosin Heavy Chain that Cause Laing Early-Onset Distal Myopathy, Affect Secondary Structure and Filament Formation In Vitro and In Vivo. J. Mol. Biol. 2018, 430, 1459–1478. [Google Scholar] [CrossRef]
- Salvi, S.S.; Kumar, R.P.; Ramachandra, N.B.; Sparrow, J.C.; Nongthomba, U. Mutations in Drosophila Myosin Rod Cause Defects in Myofibril Assembly. J. Mol. Biol. 2012, 419, 22–40. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Structural composition of betaI- and betaII-proteins. Protein Sci. 2003, 12, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, N.; Biswas, P. Position-specific propensities of amino acids in the β-strand. BMC Struct. Biol. 2010, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.K.; Withka, J.M.; Regan, L. A Thermodynamic Scale for the beta-Sheet Forming Tendencies of the Amino Acids. Biochem. 1994, 33, 5510–5517. [Google Scholar] [CrossRef]
- Eckhardt, B.; Grosse, W.; Essen, L.O.; Geyer, A. Structural characterization of a beta-turn mimic within a protein-protein interface. Proc. Natl. Acad. Sci. USA 2010, 107, 18336–18341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, J.B.; Hughes, R.A.; Alewood, P.F.; Andrews, P.R. β-turn topography. Tetrahedron 1993, 49, 3467–3478. [Google Scholar] [CrossRef]
- Blanco, F.; Ramirez-Alvarado, M.; Serrano, L. Formation and stability of beta-hairpin structures in polypeptides. Curr. Opin. Struct. Biol. 1998, 8, 107–111. [Google Scholar] [CrossRef]
- De Alba, E.; Rico, M.; Jiménez, M.A. Cross-strand side-chain interactions versus turn conformation in beta-hairpins. Protein Sci. 2008, 6, 2548–2560. [Google Scholar] [CrossRef]
- Eswar, N.; Ramakrishnan, C. Secondary structures without backbone: An analysis of backbone mimicry by polar side chains in protein structures. Protein Eng. 1999, 12, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Guruprasad, K.; Rajkumar, S. Beta-and gamma-turns in proteins revisited: A new set of amino acid turn-type dependent positional preferences and potentials. J. Biosci. 2000, 25, 143–156. [Google Scholar] [CrossRef]
- Flocco, M.M.; Mowbray, S.L. Planar Stacking Interactions of Arginine and Aromatic Side-Chains in Proteins. J. Mol. Biol. 1994, 235, 709–717. [Google Scholar] [CrossRef]
- Nishio, M.; Umezawa, Y.; Fantini, J.; Weiss, M.S.; Chakrabarti, P. CH–π hydrogen bonds in biological macromolecules. Phys. Chem. Chem. Phys. 2014, 16, 12648–12683. [Google Scholar] [CrossRef] [PubMed]
- Franklin, M.W.; Slusky, J.S. Tight Turns of Outer Membrane Proteins: An Analysis of Sequence, Structure, and Hydrogen Bonding. J. Mol. Biol. 2018, 430, 3251–3265. [Google Scholar] [CrossRef]
- Ramírez-Alvarado, M.; Blanco, F.J.; Niemann, H.; Serrano, L. Role of β-turn residues in β-hairpin formation and stability in designed peptides11Edited by A.R. Fersht. J. Mol. Biol. 1997, 273, 898–912. [Google Scholar] [CrossRef]
- Dasgupta, B.; Chakrabarti, P. pi-Turns: Types, systematics and the context of their occurrence in protein structures. BMC Struct. Biol. 2008, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, B.; Pal, L.; Basu, G.; Chakrabarti, P. Expanded turn conformations: Characterization and sequence-structure correspondence in α-turns with implications in helix folding. Proteins Struct. Funct. Bioinform. 2004, 55, 1090. [Google Scholar] [CrossRef] [Green Version]
- Craveur, P.; Joseph, A.P.; Rebehmed, J.; De Brevern, A.G. β-Bulges: Extensive structural analyses of β-sheets irregularities. Protein Sci. 2013, 22, 1366–1378. [Google Scholar] [CrossRef] [Green Version]
- Milner-White, E. Beta-bulges within loops as recurring features of protein structure. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 1987, 911, 261–265. [Google Scholar] [CrossRef]
- Khan, M.A.; Neale, C.; Michaux, C.; Pomès, R.; Privé, G.; Woody, A.R.W.; Bishop, R.E. Gauging a Hydrocarbon Ruler by an Intrinsic Exciton Probe. Biochemistry 2007, 46, 4565–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, D.; Gai, F. Exciton circular dichroism couplet arising from nitrile-derivatized aromatic residues as a structural probe of proteins. Anal. Biochem. 2016, 507, 74–78. [Google Scholar] [CrossRef] [Green Version]
- Woody, R.W. Circular Dichroism. Encycl. Mol. Biol. 2002, 246, 34–71. [Google Scholar] [CrossRef]
- Antosiewicz, J.M.; Shugar, D. UV–Vis spectroscopy of tyrosine side-groups in studies of protein structure. Part 1: Basic principles and properties of tyrosine chromophore. Biophys. Rev. 2016, 8, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Fornander, L.H.; Feng, B.; Beke-Somfai, T.; Nordén, B. UV Transition Moments of Tyrosine. J. Phys. Chem. 2014, 118, 9247–9257. [Google Scholar] [CrossRef] [PubMed]
- Noronha, M.; Lima, J.C.; Lamosa, P.; Santos, H.; Maycock, C.; Ventura, M.R.; Macanita, A. Intramolecular Fluorescence Quenching of Tyrosine by the Peptide α-Carbonyl Group Revisited. J. Phys. Chem. A 2004, 108, 2155–2166. [Google Scholar] [CrossRef]
- Brown, J.H.; Cohen, C.; Parry, D.A. Heptad Breaks in Alpha-Helical Coiled Coils: Stutters and Stammers. Proteins 1996, 26, 134–145. [Google Scholar] [CrossRef]
- Taylor, K.C.; Buvoli, M.; Korkmaz, E.N.; Buvoli, A.; Zheng, Y.; Heinze, N.T.; Cui, Q.; Leinwand, L.A.; Rayment, I. Skip residues modulate the structural properties of the myosin rod and guide thick filament assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E3806–E3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, P.M. The structure of spindle-shaped paracrystals of light meromyosin. J. Mol. Biol. 1981, 146, 201–221. [Google Scholar] [CrossRef]
- Ward, R.; Bennett, P.M. Paracrystals of myosin rod. J. Muscle Res. Cell Motil. 1989, 10, 34–52. [Google Scholar] [CrossRef]
- Franke, J.D.; Montague, R.A.; Rickoll, W.L.; Kiehart, D.P. An MYH9 human disease model in flies: Site-directed mutagenesis of the Drosophila non-muscle myosin II results in hypomorphic alleles with dominant character. Hum. Mol. Genet. 2007, 16, 3160–3173. [Google Scholar] [CrossRef] [Green Version]
- Ronen, D.; Rosenberg, M.M.; Shalev, D.E.; Rosenberg, M.; Rotem, S.; Friedler, A.; Ravid, S. The Positively Charged Region of the Myosin IIC Non-helical Tailpiece Promotes Filament Assembly. J. Biol. Chem. 2010, 285, 7079–7086. [Google Scholar] [CrossRef] [Green Version]
- Flashman, E.; Watkins, H.; Redwood, C. Localization of the binding site of the C-terminal domain of cardiac myosin-binding protein-C on the myosin rod. Biochem. J. 2006, 401, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Menard, L.; Nyland, L.; Vigoreaux, J. The Structural and Biomechanical Properties of Insect Thick Filaments Expressing Flightin and Cardiac Myosin Binding Protein-C. Microsc. Microanal. 2013, 19, 80–81. [Google Scholar] [CrossRef] [Green Version]
- Menard, L.; Wood, N.; Vigoreaux, J. Contiguity and Structural Impacts of a Non-Myosin Protein within the Thick Filament Myosin Layers. Biology 2021. submitted for publication. [Google Scholar]
- Reedy, M.C.; Beall, C. Ultrastructure of developing flight muscle in Drosophila. I. Assembly of myofibrils. Dev. Biol. 1993, 160, 443–465. [Google Scholar] [CrossRef] [PubMed]
- Vigoreaux, J.O.; Saide, J.D.; Valgeirsdottir, K.; Pardue, M.L. Flightin, a novel myofibrillar protein of Drosophila stretch-activated muscles. J. Cell Biol. 1993, 121, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Tanner, B.C.; Nyland, L.R.; Vigoreaux, J.O. Comparative biomechanics of thick filaments and thin filaments with functional consequences for muscle contraction. J. Biomed. Biotechnol. 2010, 2010, 473423. [Google Scholar] [CrossRef] [Green Version]
- Nyland, L.R.; Palmer, B.M.; Chen, Z.; Maughan, D.W.; Seidman, C.E.; Seidman, J.; Kreplak, L.; Vigoreaux, J.O. Cardiac Myosin Binding Protein-C Is Essential for Thick-Filament Stability and Flexural Rigidity. Biophys. J. 2009, 96, 3273–3280. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2° Structure | % of Total | |
|---|---|---|
| Other | 47.9 ± 1.0 | |
| Antiparallel β | Right-twisted | 23.2 ± 1.6 |
| Relaxed | 7.8 ± 1.5 | |
| Left-twisted | 0.4 ± 0.2 | |
| Turn | 14.7 ± 0.3 | |
| Helix | Alpha | 0.8 ± 0.4 |
| Distorted | 3.5 ± 1.1 | |
| Parallel β | 1.7 ± 0.8 | |
| Method | Act(sep) | Theo(sep) | Act(comb) | Theo(comb) |
|---|---|---|---|---|
| 208 nm magnitude | 45.6% | 51.6% | 23.7% | 29.8% |
| 222 nm magnitude | 24.5% | 21.5% | 16.2% | 15.1% |
| 230–240 nm slope | 24.3% | 19.3% | 10.1% | 15.4% |
| K2D | 30% | 21% | 10% | 23% |
| Contin | 14.1% | 19.7% | 9.9% | 17% |
| CDSSTR | 22% | 24% | 5% | 16.6% |
| BeStSel (190–250 nm) | 16.9% | 21.4% | 7.4% | 16.5% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menard, L.M.; Wood, N.B.; Vigoreaux, J.O. Secondary Structure of the Novel Myosin Binding Domain WYR and Implications within Myosin Structure. Biology 2021, 10, 603. https://doi.org/10.3390/biology10070603
Menard LM, Wood NB, Vigoreaux JO. Secondary Structure of the Novel Myosin Binding Domain WYR and Implications within Myosin Structure. Biology. 2021; 10(7):603. https://doi.org/10.3390/biology10070603
Chicago/Turabian StyleMenard, Lynda M., Neil B. Wood, and Jim O. Vigoreaux. 2021. "Secondary Structure of the Novel Myosin Binding Domain WYR and Implications within Myosin Structure" Biology 10, no. 7: 603. https://doi.org/10.3390/biology10070603