Genome-Wide Identification and Characterization of Long Noncoding RNAs Involved in Chinese Wheat Mosaic Virus Infection of Nicotiana benthamiana

Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Plant Growth and Virus Infection Conditions
2.2. RNA Extraction, Library Construction, and RNA-Sequencing
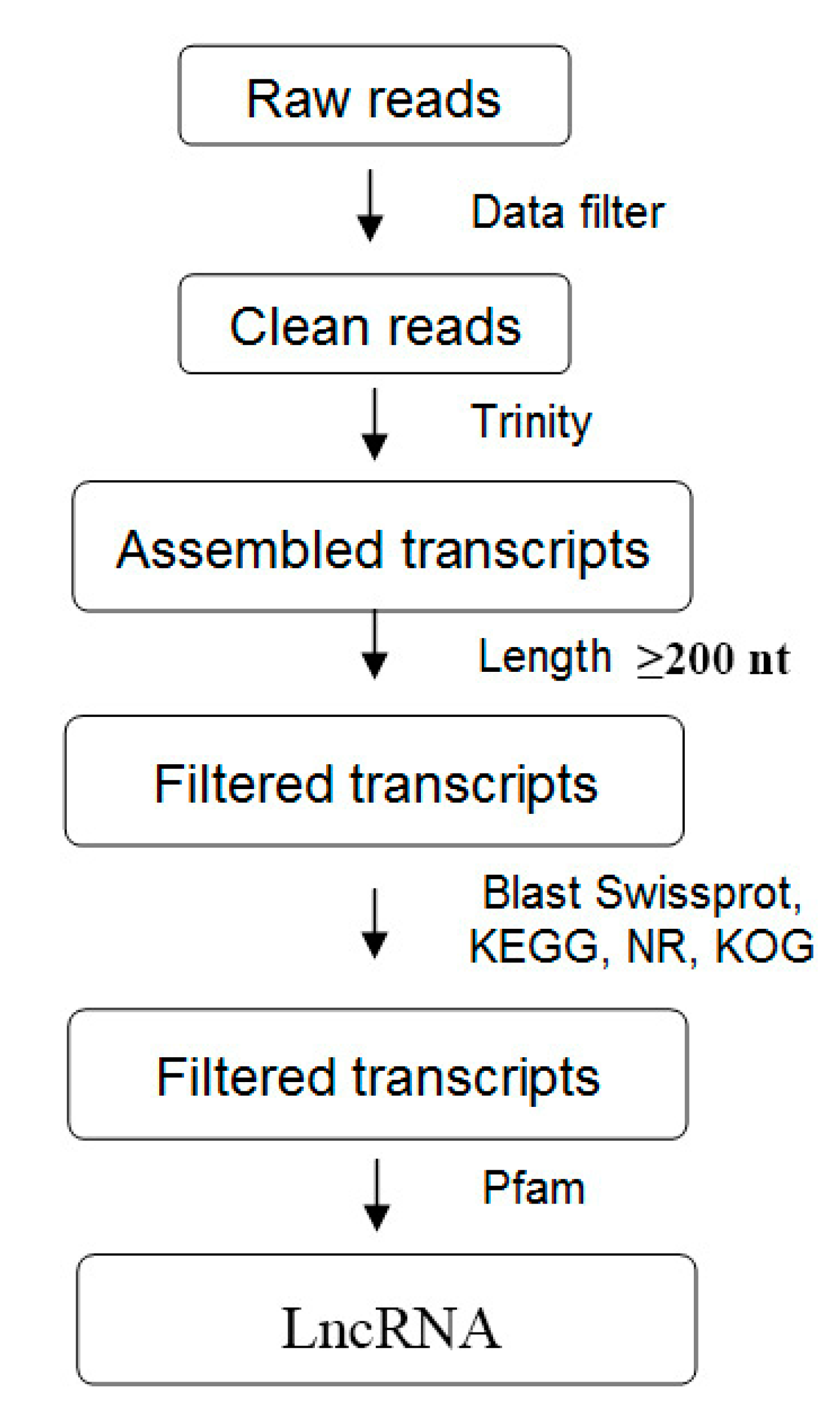
2.3. Transcript Assembly, Mapping, and Identification of LncRNAs
2.4. Analysis of Differentially Expressed Genes and Prediction of Target Genes
2.5. Construction of LncRNA, MiRNA, and MRNA Regulatory Network
2.6. Vector Construction and CWMV Infection
2.7. Protein Extraction and Western Blot Assays
3. Results
3.1. Genome-wide Identification of LncRNAs in N. benthamiana
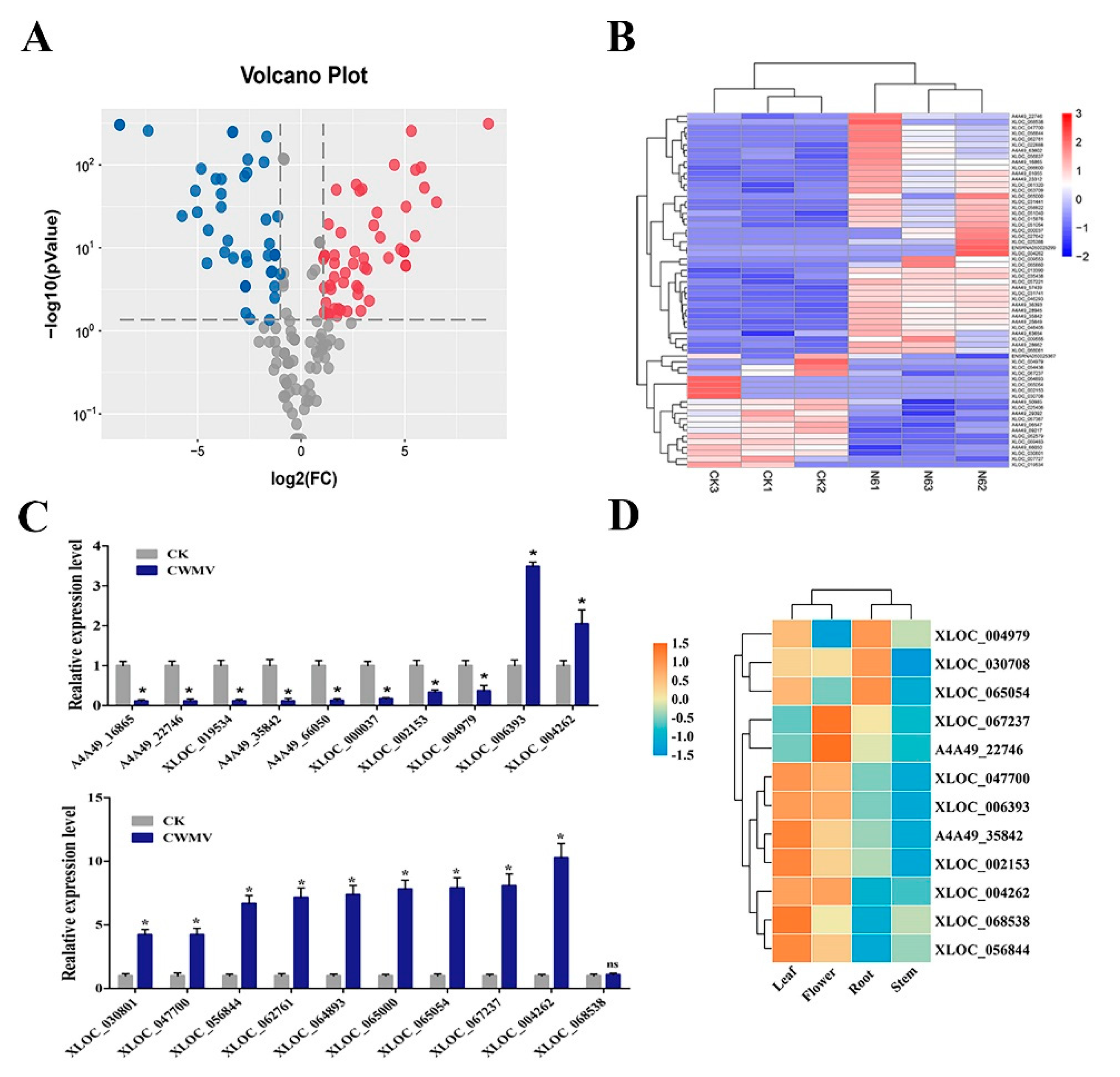
3.2. Characterization and Expression Analysis of N. benthamiana LncRNAs in Response to CWMV Infection
3.3. Identification of N. benthamiana LncRNAs by qRT-PCR
3.4. Tissue Expression Profile of N. benthamiana LncRNAs
3.5. GO and KEGG Enrichment Analysis of LncRNAs Target Genes (LTGs)
3.6. Construction lncRNA-miRNA-mRNA Regulatory Network during CWMV Infection
3.7. Silencing XLOC_006393 Inhibits Accumulation of CWMV in N. benthamiana
3.8. Expression Regulation of NbAGO1 by NbmiR168c through Post-Transcriptional Process In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, X.; Zheng, H.; Sui, N. Regulation mechanism of long non-coding RNA in plant response to stress. Biochem. Biophys. Res. Commun. 2018, 503, 402–407. [Google Scholar] [CrossRef] [PubMed]
- I H G S International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nat. Cell Biol. 2004, 431, 931–945. [CrossRef]
- Nejat, N.; Mantri, N. Emerging roles of long non-coding RNAs in plant response to biotic and abiotic stresses. Crit. Rev. Biotechnol. 2018, 38, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Ariel, F.; Romero-Barrios, N.; Jégu, T.; Benhamed, M.; Crespi, M. Battles and hijacks: Noncoding transcription in plants. Trends Plant Sci. 2015, 20, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, C.; Bao, H.; Chen, H.; Wang, Y. Genome-wide identification and characterization of novel lncRNAs in Populus under nitrogen deficiency. Mol. Genet. Genom. 2016, 291, 1663–1680. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.-H. Genome-Wide Analysis Uncovers Regulation of Long Intergenic Noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Sun, S.; Li, P.; Bu, D.; Cao, H.; Zhao, Y. Comprehensive Characterization of 10,571 Mouse Large Intergenic Noncoding RNAs from Whole Transcriptome Sequencing. PLoS ONE 2013, 8, e70835. [Google Scholar] [CrossRef]
- Zhou, Z.-Y.; Li, A.-M.; Adeola, A.C.; Liu, Y.-H.; Irwin, D.M.; Xie, H.-B.; Zhang, Y.-P. Genome-Wide Identification of Long Intergenic Noncoding RNA Genes and Their Potential Association with Domestication in Pigs. Genome Biol. Evol. 2014, 6, 1387–1392. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.-C.; Kean, M.J.; Xu, J.; Chua, N.-H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-C.; Liao, J.-Y.; Li, Z.-Y.; Yu, Y.; Zhang, J.-P.; Li, Q.-F.; Qu, L.-H.; Shu, W.-S.; Chen, Y.-Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ai, G.; Zhang, C.; Cui, L.; Wang, J.; Li, H.; Zhang, J.; Ye, Z. Expression and diversification analysis reveals transposable elements play important roles in the origin of Lycopersicon-specific lncRNAs in tomato. New Phytol. 2015, 209, 1442–1455. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R.; Steitz, J.A. The Noncoding RNA Revolution—Trashing Old Rules to Forge New Ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [Green Version]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Gloss, B.S.; Dinger, M.E. The specificity of long noncoding RNA expression. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1859, 16–22. [Google Scholar] [CrossRef]
- Zhu, Q.-H.; Stephen, S.; Taylor, J.; Helliwell, C.A.; Wang, M.-B. Long noncoding RNAs responsive toFusarium oxysporuminfection inArabidopsis thaliana. New Phytol. 2014, 201, 574–584. [Google Scholar] [CrossRef]
- Ding, J.; Shen, J.; Mao, H.; Xie, W.; Li, X.; Zhang, Q. RNA-Directed DNA Methylation Is Involved in Regulating Photoperiod-Sensitive Male Sterility in Rice. Mol. Plant 2012, 5, 1210–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Hao, L.; Li, D.; Zhu, L.; Hu, S. Long Non-coding RNAs and Their Biological Roles in Plants. Genom. Proteom. Bioinform. 2015, 13, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Niu, Q.-W.; Wu, H.-W.; Liu, J.; Ye, J.; Yu, N.; Chua, N.-H. Analysis of non-coding transcriptome in rice and maize uncovers roles of conserved lncRNAs associated with agriculture traits. Plant J. 2015, 84, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Marquardt, S.; Lister, C.; Swiezewski, S.; Dean, C. Targeted 3′ Processing of Antisense Transcripts Triggers Arabidopsis FLC Chromatin Silencing. Science 2009, 327, 94–97. [Google Scholar] [CrossRef]
- Tian, Y.; Bai, S.; Dang, Z.; Hao, J.; Zhang, J.; Hasi, A. Genome-wide identification and characterization of long non-coding RNAs involved in fruit ripening and the climacteric in Cucumis melo. BMC Plant Biol. 2019, 19, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.B.; Sung, S. Vernalization-Mediated Epigenetic Silencing by a Long Intronic Noncoding RNA. Science 2010, 331, 76–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zheng, Y.; Ham, B.; Zhang, S.; Fei, Z.; Lucas, W.J. Plant lncRNAs are enriched in and move systemically through the phloem in response to phosphate deficiency. J. Integr. Plant Biol. 2019, 61, 492–508. [Google Scholar] [CrossRef]
- Wang, J.; Yu, W.; Yang, Y.; Li, X.; Chen, T.; Liu, T.; Ma, N.; Yang, X.; Liu, R.; Zhang, B. Genome-wide analysis of tomato long non-coding RNAs and identification as endogenous target mimic for microRNA in response to TYLCV infection. Sci. Rep. 2015, 5, 16946. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Hu, W.; Hao, J.; Lv, S.; Wang, C.; Tong, W.; Wang, Y.; Wang, Y.; Liu, X.; Ji, W. Genome-wide identification and functional prediction of novel and fungi-responsive lincRNAs in Triticum aestivum. BMC Genom. 2016, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Jiang, N.; Meng, J.; Yang, G.; Liu, W.; Zhou, X.; Ma, N.; Hou, X.; Luan, Y. LncRNA33732-respiratory burst oxidase module associated with WRKY1 in tomato- Phytophthora infestans interactions. Plant J. 2018, 97, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, T.; Shen, D.; Wang, J.; Ling, X.; Hu, Z.; Chen, T.; Hu, J.; Huang, J.; Yu, W.; et al. Tomato yellow leaf curl virus intergenic siRNAs target a host long noncoding RNA to modulate disease symptoms. PLoS Pathog. 2019, 15, e1007534. [Google Scholar] [CrossRef]
- Adams, M.J.; Antoniw, J.F.; Kreuze, J. Virgaviridae: A new family of rod-shaped plant viruses. Arch. Virol. 2009, 154, 1967–1972. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, T.-Y.; Liao, Q.-S.; He, L.; Li, J.; Zhang, H.-M.; Chen, X.; Li, J.; Li, J.-B.; Chen, J.-P.; et al. Chinese Wheat Mosaic Virus-Induced Gene Silencing in Monocots and Dicots at Low Temperature. Front. Plant Sci. 2018, 9, 1627. [Google Scholar] [CrossRef]
- Andika, I.B.; Zheng, S.; Tan, Z.; Sun, L.; Kondo, H.; Zhou, X.; Chen, J. Endoplasmic reticulum export and vesicle formation of the movement protein of Chinese wheat mosaic virus are regulated by two transmembrane domains and depend on the secretory pathway. Virology 2013, 435, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Andika, I.B.; Kondo, H.; Chen, J. Identification of the amino acid residues and domains in the cysteine-rich protein ofChinese wheat mosaic virusthat are important for RNA silencing suppression and subcellular localization. Mol. Plant Pathol. 2013, 14, 265–278. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, F.; Xie, L.; Song, X.-J.; Li, J.; Chen, J.-P.; Zhang, H.-M. Functional identification of two minor capsid proteins from Chinese wheat mosaic virus using its infectious full-length cDNA clones. J. Gen. Virol. 2016, 97, 2441–2450. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009, 23, 205–211. [Google Scholar]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Burge, S.W.; Daub, J.; Eberhardt, R.Y.; Tate, J.G.; Barquist, L.; Nawrocki, E.P.; Eddy, S.R.; Gardner, P.P.; Bateman, A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2012, 41, D226–D232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Joseph, Z.; Gifford, D.K.; Jaakkola, T.S. Fast optimal leaf ordering for hierarchical clustering. Bioinformatics 2001, 17, S22–S29. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Luan, Y.; Jiang, N.; Bao, H.; Meng, J. Comparative transcriptome analysis between resistant and susceptible tomato allows the identification of lncRNA16397 conferring resistance toPhytophthora infestansby co-expressing glutaredoxin. Plant J. 2017, 89, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Karakülah, G.; Kurtoğlu, K.Y.; Unver, T. PeTMbase: A Database of Plant Endogenous Target Mimics (eTMs). PLoS ONE 2016, 11, e0167698. [Google Scholar] [CrossRef]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.-Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Shen, A.; Liu, A. Long non-coding RNA H19 and cancer: A competing endogenous RNA. Bull Cancer 2019, 106, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, L.; Zhu, B.; Zhu, H.; Luo, Y.; Wang, Q.; Zuo, J. Integrative analysis of long non-coding RNA acting as ceRNAs involved in chilling injury in tomato fruit. Gene 2018, 667, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Gursinsky, T.; Pirovano, W.; Gambino, G.; Friedrich, S.; Behrens, S.-E.; Pantaleo, V. Homeologs of the Nicotiana benthamiana Antiviral ARGONAUTE1 Show Different Susceptibilities to microRNA168-Mediated Control. Plant Physiol. 2015, 168, 938–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Várallyay, É.; Válóczi, A.; Ágyi, Á.; Burgyán, J.; Havelda, Z. Plant virus-mediated induction of miR168 is associated with repression of ARGONAUTE1 accumulation. EMBO J. 2010, 29, 3507–3519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Huang, Y.; Yang, J.; Yao, S.; Zhao, K.; Wang, D.; Qin, Q.; Bian, Z.; Li, Y.; Lan, Y.; et al. Jasmonate Signaling Enhances RNA Silencing and Antiviral Defense in Rice. Cell Host Microbe 2020, 28, 89–103.e8. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Reads | Clean Reads | Clean Reads Rate (%) | Q20% | Q30% | GC% | Mapped Reads | Mapped Reads Rate (%) |
|---|---|---|---|---|---|---|---|---|
| CK1 | 98,235,824 | 96,516,697 | 98.25 | 97.06 | 93.87 | 43.95 | 59,010,308 | 61.14 |
| CK2 | 104,512,954 | 103,132,920 | 98.68 | 97.14 | 94.04 | 44.85 | 61,372,953 | 59.51 |
| CK3 | 107,653,544 | 106,146,914 | 98.60 | 97.22 | 94.24 | 43.74 | 63,872,643 | 60.17 |
| N61 | 101,064,306 | 99,414,122 | 98.37 | 97.16 | 94.08 | 43.95 | 49,184,563 | 49.47 |
| N62 | 95,583,802 | 93,815,501 | 98.15 | 97.26 | 94.27 | 43.78 | 47,780,234 | 50.93 |
| N63 | 95,751,460 | 94,688,618 | 98.89 | 96.25 | 92.2 | 43.79 | 52,078,740 | 55.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, W.; Hu, H.; Lu, Q.; Jin, P.; Cai, L.; Hu, C.; Yang, J.; Dai, L.; Chen, J. Genome-Wide Identification and Characterization of Long Noncoding RNAs Involved in Chinese Wheat Mosaic Virus Infection of Nicotiana benthamiana. Biology 2021, 10, 232. https://doi.org/10.3390/biology10030232
Zheng W, Hu H, Lu Q, Jin P, Cai L, Hu C, Yang J, Dai L, Chen J. Genome-Wide Identification and Characterization of Long Noncoding RNAs Involved in Chinese Wheat Mosaic Virus Infection of Nicotiana benthamiana. Biology. 2021; 10(3):232. https://doi.org/10.3390/biology10030232
Chicago/Turabian StyleZheng, Weiran, Haichao Hu, Qisen Lu, Peng Jin, Linna Cai, Cailin Hu, Jian Yang, Liangying Dai, and Jianping Chen. 2021. "Genome-Wide Identification and Characterization of Long Noncoding RNAs Involved in Chinese Wheat Mosaic Virus Infection of Nicotiana benthamiana" Biology 10, no. 3: 232. https://doi.org/10.3390/biology10030232