
First-Principles Calculations to Investigate the Oxidation Mechanism of Pristine MoS2 and Ti-Doped MoS2



Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Phonon Dispersion
3.2. Adsorption Energy
3.3. Charge Density Difference
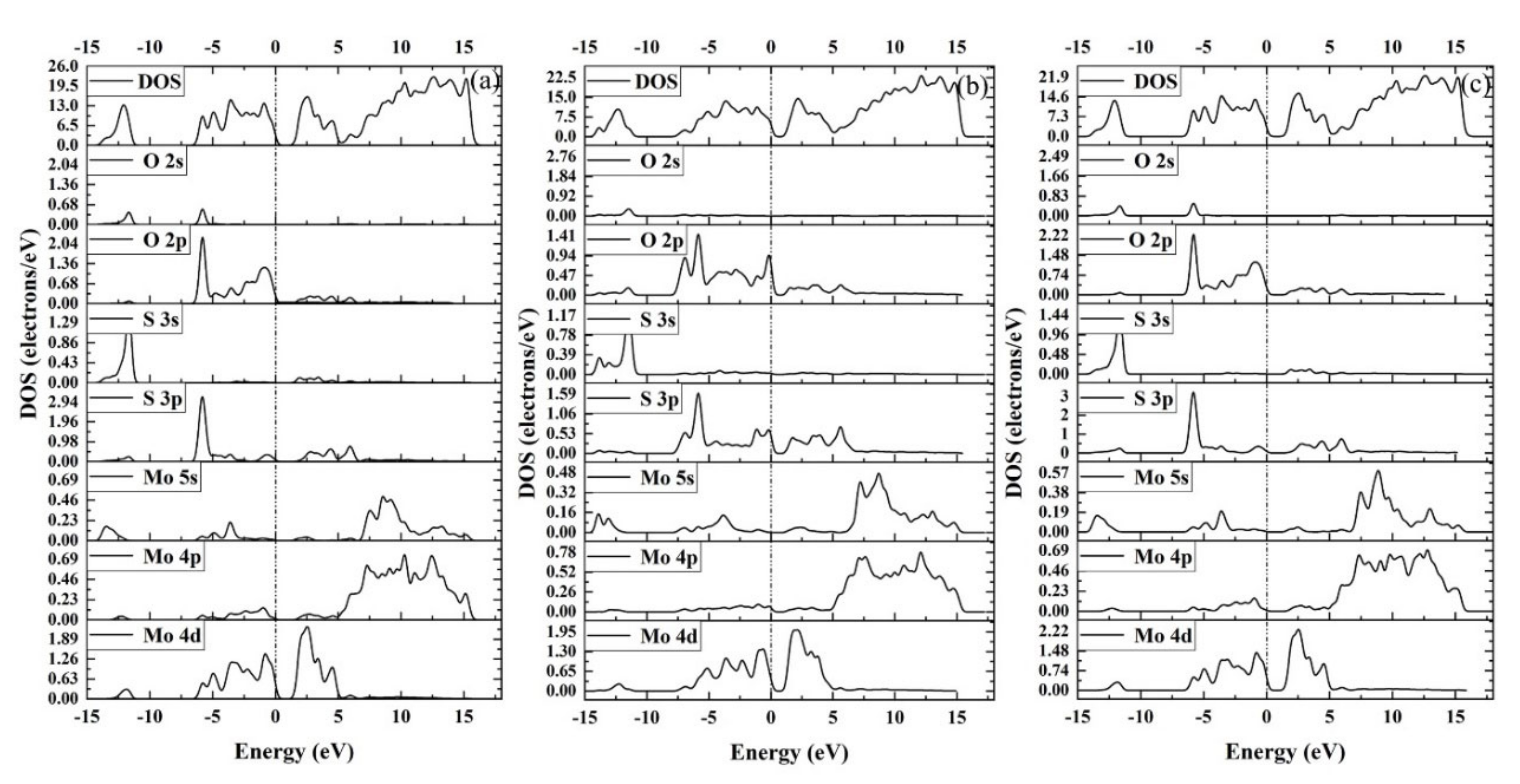
3.4. Density of States (DOS)
4. Conclusions
- (1)
- The oxidation mechanism of the pristine MoS2 was well probed. Two Ti-doped models, MSTi-S and MSTi-Mo, were designed based on the structural feature of MoS2. Due to the dynamical instability of MSTi-S, the antioxidation behavior of MSTi-Mo was investigated in detail.
- (2)
- Although the antioxidation property of the Mo site improved, the H site and Ti top site were easily attacked by the O atom, leading to broken Ti–S bonds. The incorporation of the Ti element into the MoS2 was not helpful for improving its antioxidation performance.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, Y.H.; Zhang, X.Q.; Zhang, W.; Chang, M.T.; Lin, C.T.; Chang, K.D.; Yu, Y.C.; Wang, J.T.W.; Chang, C.S.; Li, L.J.; et al. Synthesis of large-area MoS2 atomic layers with chemical vapor deposition. Adv. Mater. 2012, 24, 2320–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimi, M.; Shokrgozar, M.A.; Hamid, D.H.; Vossoughi, M. Photothermal properties of two-dimensional molybdenum disulfide (MoS2) with nanoflower and nanosheet morphology. Mater. Res. Bull. 2022, 152, 111837. [Google Scholar] [CrossRef]
- Huang, W.; Li, Y.; Fu, Q.; Chen, M. Fabrication of a novel biochar decorated nano-flower-like MoS2 nanomaterial for the enhanced photodegradation activity of ciprofloxacin: Performance and mechanism. Mater. Res. Bull. 2022, 147, 111650. [Google Scholar] [CrossRef]
- Uddin, M.K.; Mashkoor, F.; Al Arifi, I.M.; Nasar, A. Simple one-step synthesis process of novel MoS2@bentonite magnetic nanocomposite for efficient adsorption of crystal violet from aqueous solution. Mater. Res. Bull. 2021, 139, 111279. [Google Scholar] [CrossRef]
- Tan, X.; Duan, Z.; Liu, H.; Wu, X.; Cho, Y.R. Core-shell structured MoS2/Ni9S8 electrocatalysts for high performance hydrogen and oxygen evolution reactions. Mater. Res. Bull. 2022, 146, 111626. [Google Scholar] [CrossRef]
- Krishnan, U.; Kaur, M.; Kaur, G.; Singh, K.; Dogra, A.R.; Kumar, M.; Kumar, A. MoS2/ZnO nanocomposites for efficient photocatalytic degradation of industrial pollutants. Mater. Res. Bull. 2019, 111, 212–221. [Google Scholar] [CrossRef]
- Imani Yengejeh, S.; Liu, J.; Kazemi, S.A.; Wen, W.; Wang, Y. Effect of structural phases on mechanical properties of molybdenum disulfide. ACS Omega 2020, 5, 5994–6002. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Gao, S.; Chen, J.; Peng, L.; Liu, K.; Wei, X. Superlubricity between MoS2 monolayers. Adv. Mtaer. 2017, 29, 1701474. [Google Scholar] [CrossRef]
- Ren, S.; Li, H.; Cui, M.; Wang, L.; Pu, J. Functional regulation of Pb-Ti/MoS2 composite coatings for environmentally adaptive solid lubrication. Appl. Surf. Sci. 2017, 401, 362–372. [Google Scholar] [CrossRef]
- Li, Y.; Xie, M.; Sun, Q.; Xu, X.; Fan, X.; Zhang, G.; Li, H.; Zhu, M. The effect of atmosphere on the tribological behavior of magnetron sputtered MoS2 coatings. Surf. Coat. Technol. 2019, 378, 125081. [Google Scholar] [CrossRef]
- Le, D.; Rawal, T.B.; Rahman, T.S. Single-layer MoS2 with sulfur vacancies: Structure and catalytic application. J. Phys. Chem. C 2014, 118, 5346–5351. [Google Scholar] [CrossRef]
- Ren, S.M.; Cui, M.J.; Martini, A.; Shi, Y.B.; Wang, H.X.; Pu, J.B.; Li, Q.Y.; Wang, L.P. Macroscale superlubricity enabled by rationally designed MoS2-based superlattice films. Res. Sq. 2022. preprint. [Google Scholar] [CrossRef]
- Santosh, K.; Roberto, C.; Robert, M.; Kyeongjae, C. Surface oxidation energetics and kinetics on MoS2 monolayer. J. Appl. Phys. 2015, 117, 136301. [Google Scholar]
- Sen, H.S.; Sahin, H.; Peeters, F.M.; Durgun, E. Monolayers of MoS2 as an oxidation protective nanocoating material. J. Appl. Phys. 2014, 116, 083508. [Google Scholar] [CrossRef] [Green Version]
- Ataca, C.; Ciraci, S. Dissociation of H2O at the vacancies of single-layer MoS2. Phys. Rev. B 2012, 85, 195410. [Google Scholar] [CrossRef]
- Li, H.; Huang, M.; Cao, G. Markedly different adsorption behaviors of gas molecules on defective monolayer MoS2: A first-principles study. Phys. Chem. Chem. Phys. 2016, 18, 15110–15117. [Google Scholar] [CrossRef]
- Li, Q.; Zheng, S.; Pu, J.; Wang, W.; Li, L.; Wang, L. Revealing the failure mechanism and designing protection approach for MoS2 in humid environment by first-principles investigation. Appl. Surf. Sci. 2019, 487, 1121–1130. [Google Scholar] [CrossRef]
- Chen, Z.W.; Yan, J.M.; Zheng, W.T.; Jiang, Q. Cu4 cluster doped monolayer MoS2 for CO oxidation. Sci. Rep. 2015, 5, 11230. [Google Scholar] [CrossRef] [Green Version]
- Cong, W.T.; Tang, Z.; Zhao, X.G.; Chu, J.H. Enhanced magnetic anisotropies of single transition-metal adatoms on a defective MoS2 nonolayer. Sci. Rep. 2015, 5, 9361. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.; Pu, J.; Wang, H.; Zheng, S.; Wang, L.; Xue, Q. Study on atmospheric tribology performance of MoS2–W films with self-adaption to temperature. Ceram. Int. 2019, 45, 15834–15842. [Google Scholar] [CrossRef]
- Li, L.; Lu, Z.; Pu, J.; Wang, H.; Li, Q.; Chen, S.; Zhang, Z.; Wang, L. The superlattice structure and self-adaptive performance of C–Ti/MoS2 composite coatings. Ceram. Int. 2020, 46, 5733–5744. [Google Scholar] [CrossRef]
- Zeng, C.; Pu, J.; Wang, H.; Zheng, S.; Chen, R. Influence of microstructure on tribological properties and corrosion resistance of MoS2/WS2 films. Ceram. Int. 2020, 46, 13774–13783. [Google Scholar] [CrossRef]
- Li, H.; Xie, M.; Zhang, G.; Fan, X.; Li, X.; Zhu, M.; Wang, L. Structure and tribological behavior of Pb-Ti/MoS2 nanoscaled multilayer films deposited by magnetron sputtering method. Appl. Surf. Sci. 2018, 435, 48–54. [Google Scholar] [CrossRef]
- Wang, P.; Yue, W.; Lu, Z.; Zhang, G.; Zhu, L. Friction and wear properties of MoS2-based coatings sliding against Cu and Al under electric current. Tribol. Int. 2018, 127, 379–388. [Google Scholar] [CrossRef]
- Lu, X.; Yan, M.; Yan, Z.; Chen, W.; Sui, X.; Hao, J.; Liu, W. Exploring the atmospheric tribological properties of MoS2-(Cr, Nb, Ti, Al, V) composite coatings by high throughput preparation method. Tribol. Int. 2021, 156, 106844. [Google Scholar] [CrossRef]
- Pan, Y. Influence of N-vacancy on the electronic and optical properties of bulk GaN from first-principles investigations. Int. J. Energy Res. 2021, 45, 15512–15520. [Google Scholar] [CrossRef]
- Chen, S.; Pan, Y. Influence of group III and IV elements on the hydrogen evolution reaction of MoS2 disulfide. J. Phys. Chem. C 2021, 125, 11848–11856. [Google Scholar] [CrossRef]
- Zhang, R.; Lu, Z.; Yang, Y.; Shi, W. First-principles investigation of the monoclinic NaMnO2 cathode material for rechargeable Na-ion batteries. Curr. Appl. Phys. 2018, 18, 1431–1435. [Google Scholar] [CrossRef]
- Zhang, R.; Zhao, J.; Yang, Y.; Shi, W.; Lu, Z.; Wang, J. Understanding the friction behavior of sulfur-terminated diamond-like carbon films under high vacuum by first-principles calculations. Curr. Appl. Phys. 2018, 18, 317–323. [Google Scholar] [CrossRef]
- Zhang, R.; Zhao, J.; Pu, J.; Lu, Z. First-Principles Investigation on the Tribological Properties of h-BN Bilayer Under Variable Load. Tribol. Lett. 2018, 66, 124. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, S.; Mao, P.; Jin, C. Role of Si concentration on the thermodynamic properties of molybdenum silicides. Vacuum 2017, 141, 170–172. [Google Scholar] [CrossRef]
- Pan, Y. The structural, mechanical and thermodynamic properties of the orthorhombic TMAl (TM=Ti, Y, Zr and Hf) aluminides from first-principles calculations. Vacuum 2020, 181, 109742. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, E. First-principles investigation of electronic and optical properties of H-doped FeS2. Int. J. Energy Res. 2021, 45, 11284–11293. [Google Scholar] [CrossRef]
- Pan, Y. First-principles investigation of the structural, mechanical, and thermodynamic properties of hexagonal and cubic MoAl5 alloy. J. Mater. Eng. Perform. 2021, 30, 8289–8295. [Google Scholar] [CrossRef]
- Tornatzky, H.; Gillen, R.; Uchiyama, H.; Maultzsch, J. Phonon dispersion in MoS2. Phys. Rev. B 2019, 99, 144309. [Google Scholar] [CrossRef] [Green Version]
- Pu, D.; Pan, Y. First-principles investigation of oxidation mechanism of Al-doped Mo5Si3 silicide. Ceram. Int. 2022, 48, 11518–11526. [Google Scholar] [CrossRef]
- Williamson, I.; Li, S.; Correa Hernandez, A.; Lawson, M.; Chen, Y.; Li, L. Structural, electrical, phonon, and optical properties of Ti- and V-doped two-dimensional MoS2. Chem. Phys. Lett. 2017, 674, 157–163. [Google Scholar] [CrossRef]
- Pan, Y.; Wen, M. Insight into the oxidation mechanism of Nb3Si (111) surface: First-principles calculations. Mater. Res. Bull. 2018, 107, 484–491. [Google Scholar] [CrossRef]
- Das, N.K.; Shoji, T. Early-stage oxidation of Ni–Cr binary alloy (111), (110) and (100) surfaces: A combined density functional and quantum chemical molecular dynamics study. Corros. Sci. 2013, 73, 18–31. [Google Scholar] [CrossRef]
- Elgendy, A.; Elkholy, A.E.; El Basiony, N.M.; Migahed, M.A. Monte Carlo simulation for the antiscaling performance of Gemini ionic liquids. J. Mol. Liq. 2019, 285, 408–415. [Google Scholar] [CrossRef]
- Guo, L.; Tan, J.; Kaya, S.; Leng, S.; Li, Q.; Zhang, F. Multidimensional insights into the corrosion inhibition of 3,3-dithiodipropionic acid on Q235 steel in H2SO4 medium: A combined experimental and in silico investigation. J. Colloid Interface Sci. 2020, 570, 116–124. [Google Scholar] [CrossRef]
- Islam, Z.; Haque, A. Defects and grain boundary effects in MoS2: A molecular dynamics study. J. Phys. Chem. Solids 2021, 148, 109669. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Q.; Liang, J.; Li, Q.; Dai, J.; Li, W. Optical properties of N and transition metal R (R=V, Cr, Mn, Fe, Co, Ni, Cu, and Zn) codoped anatase TiO2. Phys. B Condens. Matter 2012, 407, 2709–2715. [Google Scholar] [CrossRef]
- Michalczyk, M.; Zierkiewicz, W.; Scheiner, S. Interactions of (MY)6 (M=Zn, Cd; Y=O, S, Se) quantum dots with N-bases. Struct. Chem. 2019, 30, 1003–1014. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Z.; Peng, X.; Zhong, J. Tuning the Dirac cone of the topological insulator Bi2Te3 thin films by substitutional nonmagnetic atoms. Phys. B Condens. Matter 2015, 456, 355–358. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Configurations | Adsorbed Site | (eV) | dS−O (Å) | dMo−O (Å) | dTi−O (Å) | dTi−Mo (Å) |
|---|---|---|---|---|---|---|
| MoS2 | H | −5.53 | 1.484 | - | - | - |
| Mo top | −3.96 | 1.568 | 2.204 | - | - | |
| S top | −5.52 | 1.483 | - | - | - | |
| Ti−MoS2 | H | −1.33 | 1.639 | 2.336 | 2.010 | - |
| Mo top | −0.70 | 1.578 | - | - | - | |
| S top | −1.90 | 1.487 | - | - | - | |
| Ti top | −1.04 | 1.601 | - | 2.062 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leng, S.; Zhang, Q.; Guo, L.; Huang, Y.; Ebenso, E.E.; Marzouki, R. First-Principles Calculations to Investigate the Oxidation Mechanism of Pristine MoS2 and Ti-Doped MoS2. Coatings 2022, 12, 1114. https://doi.org/10.3390/coatings12081114
Leng S, Zhang Q, Guo L, Huang Y, Ebenso EE, Marzouki R. First-Principles Calculations to Investigate the Oxidation Mechanism of Pristine MoS2 and Ti-Doped MoS2. Coatings. 2022; 12(8):1114. https://doi.org/10.3390/coatings12081114
Chicago/Turabian StyleLeng, Senlin, Qiao Zhang, Lei Guo, Yue Huang, Eno E. Ebenso, and Riadh Marzouki. 2022. "First-Principles Calculations to Investigate the Oxidation Mechanism of Pristine MoS2 and Ti-Doped MoS2" Coatings 12, no. 8: 1114. https://doi.org/10.3390/coatings12081114