
First-Principles Study on the Adsorption and Dissociation Behavior of H2 on the Surface of a Plutonium–Gallium System

Abstract
:1. Introduction
2. Calculation Methods and Models
2.1. Calculation Method
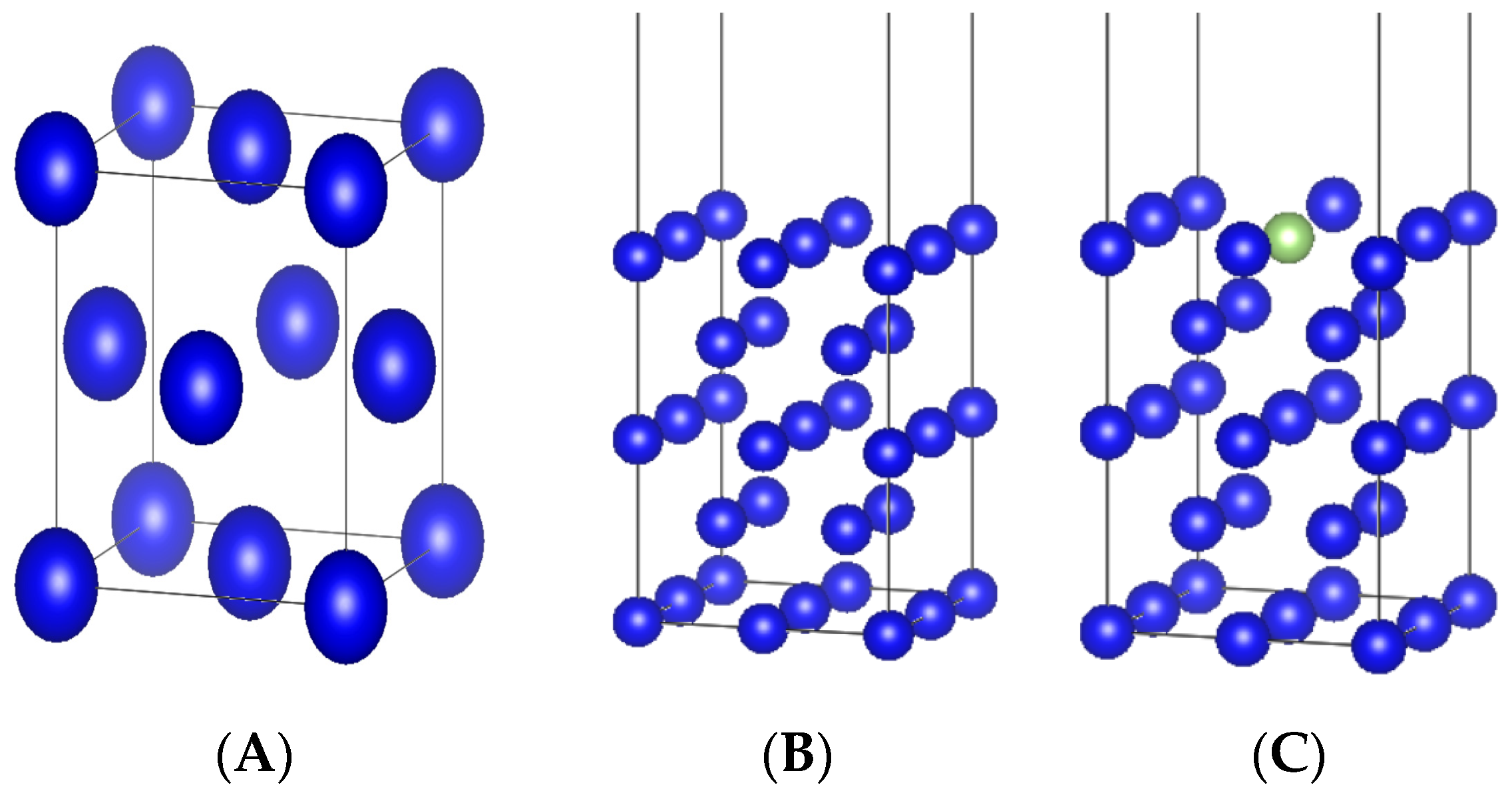
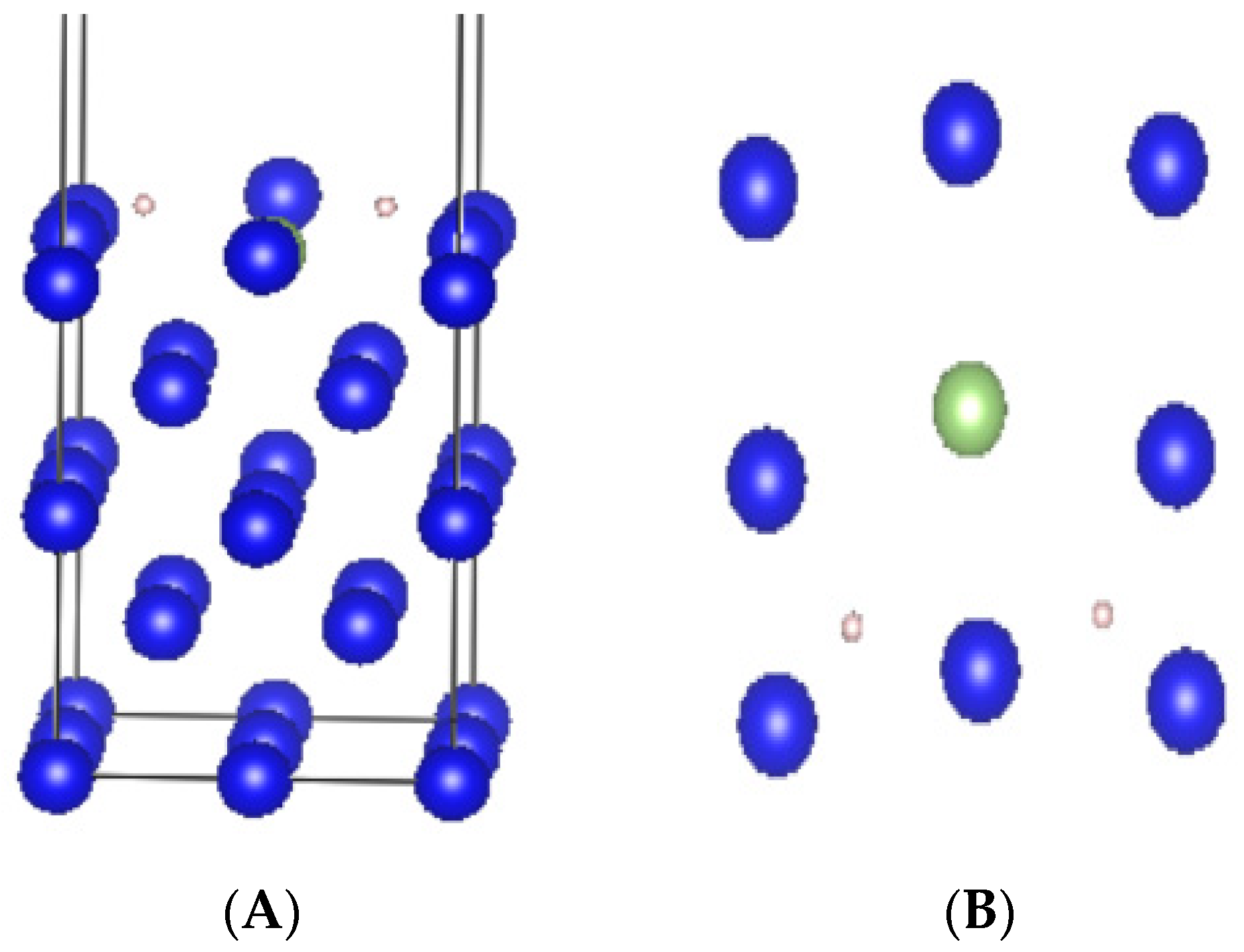
2.2. Computational Models
3. Results and Discussion
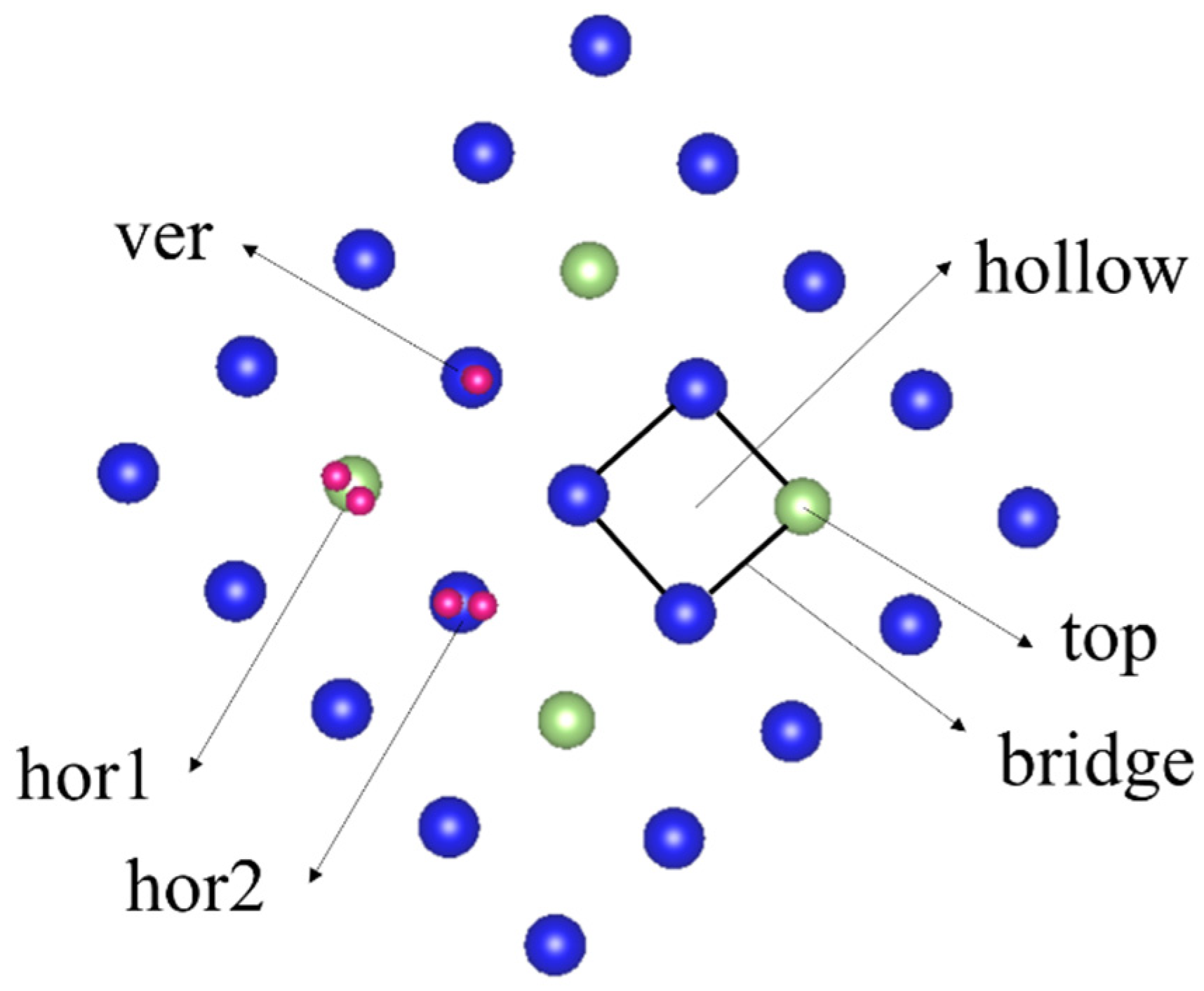
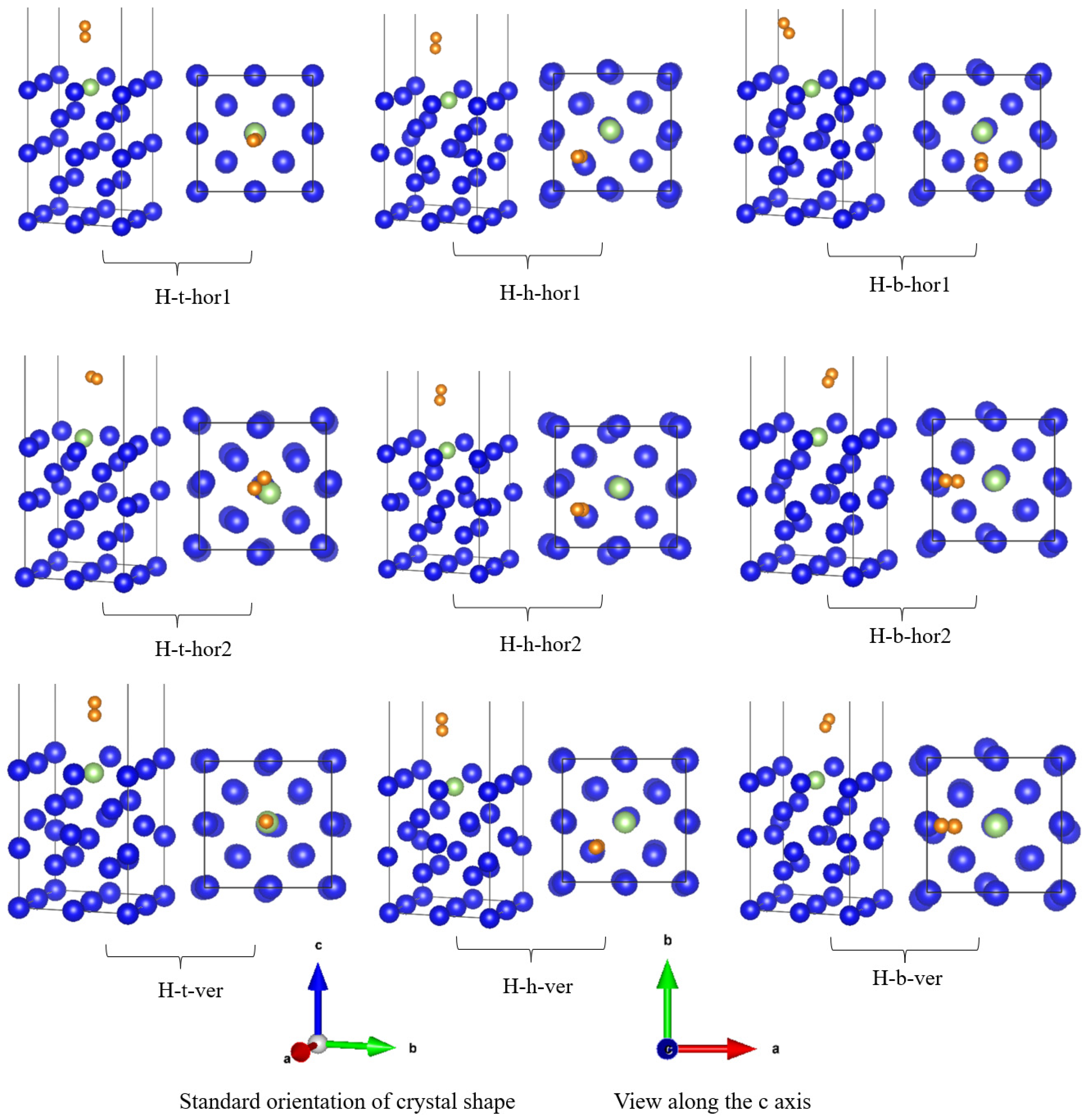
3.1. Adsorption Conformation and Adsorption Energy
3.2. Bader Charge Analysis
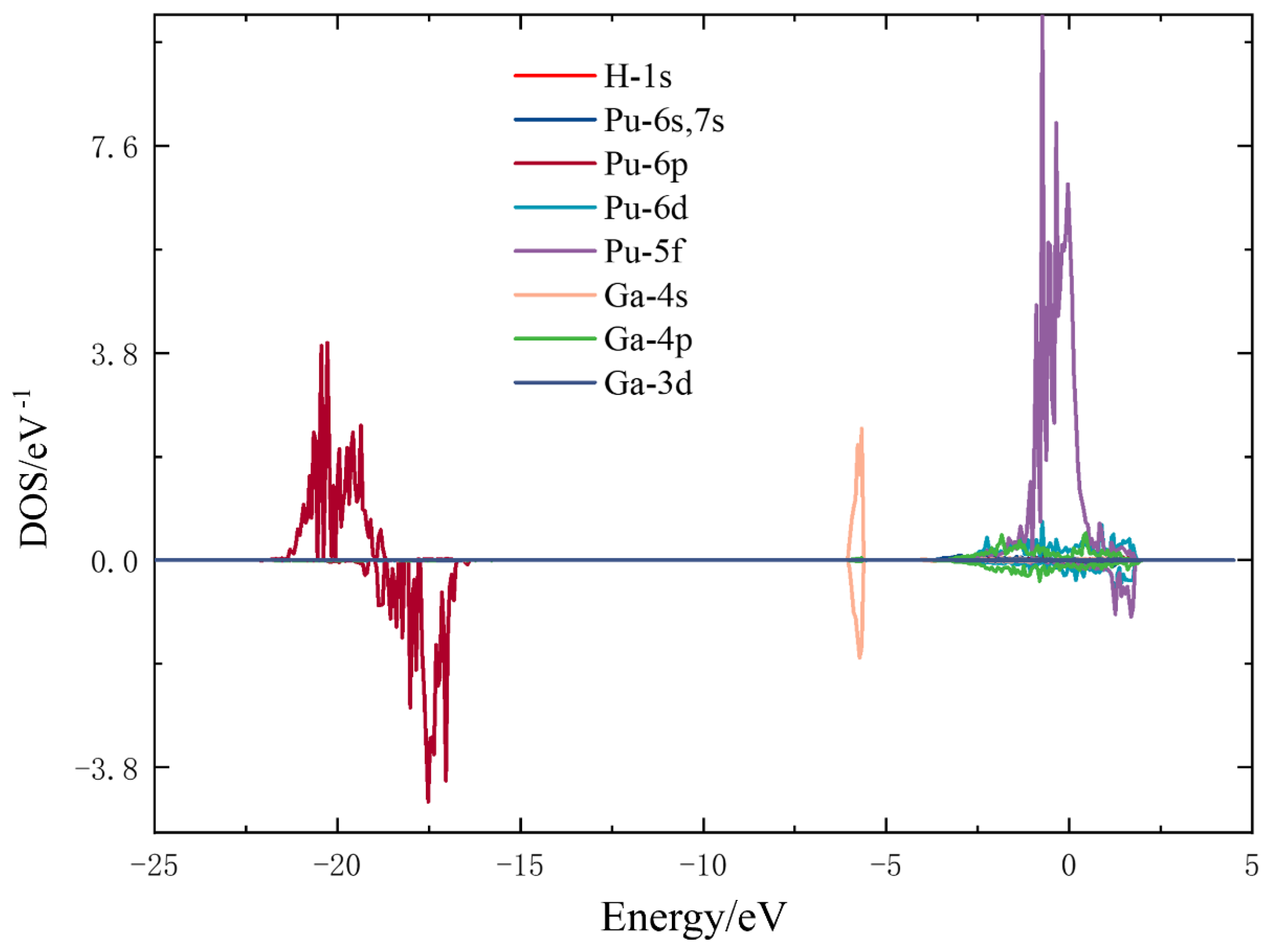
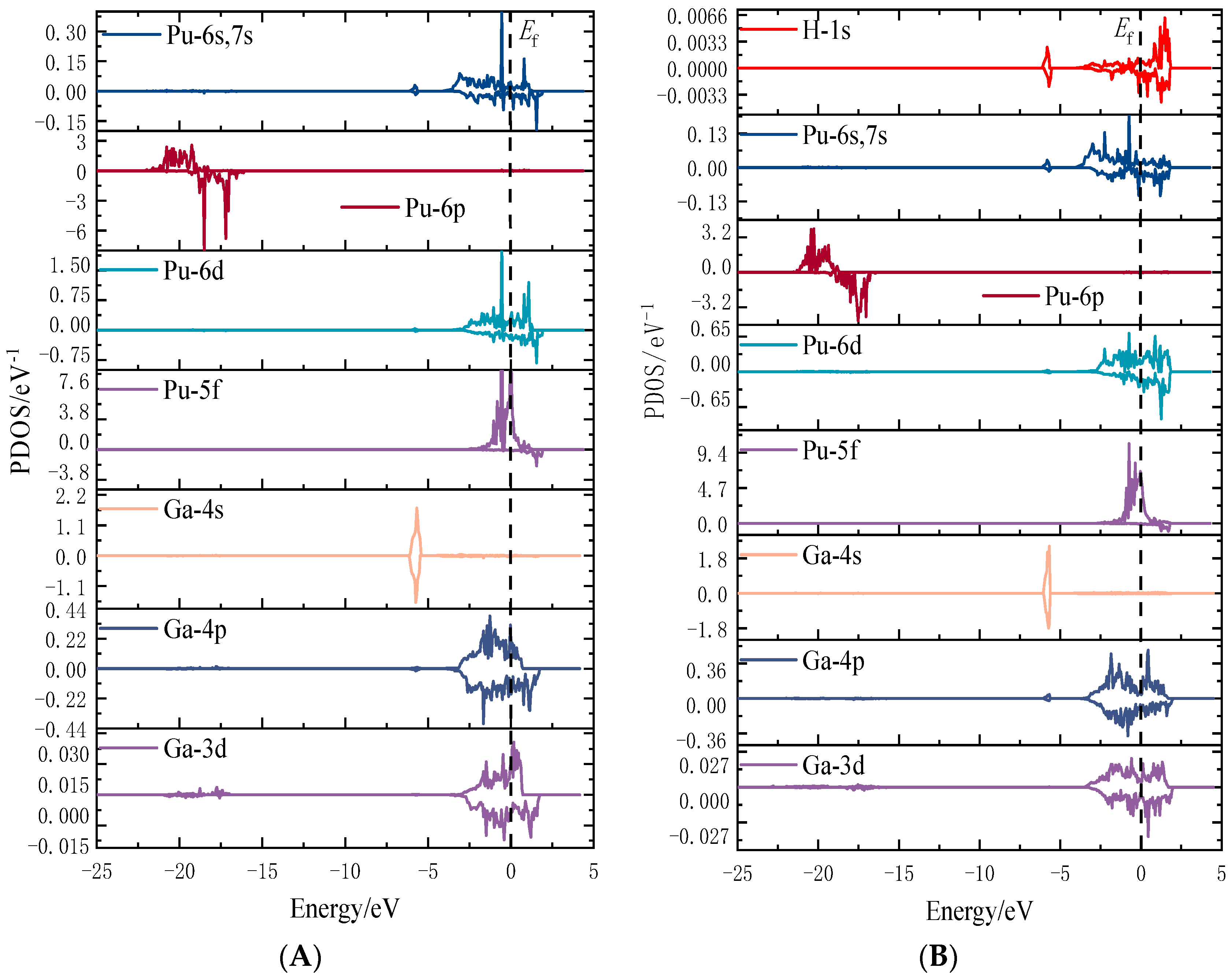
3.3. Electronic Structure Analysis
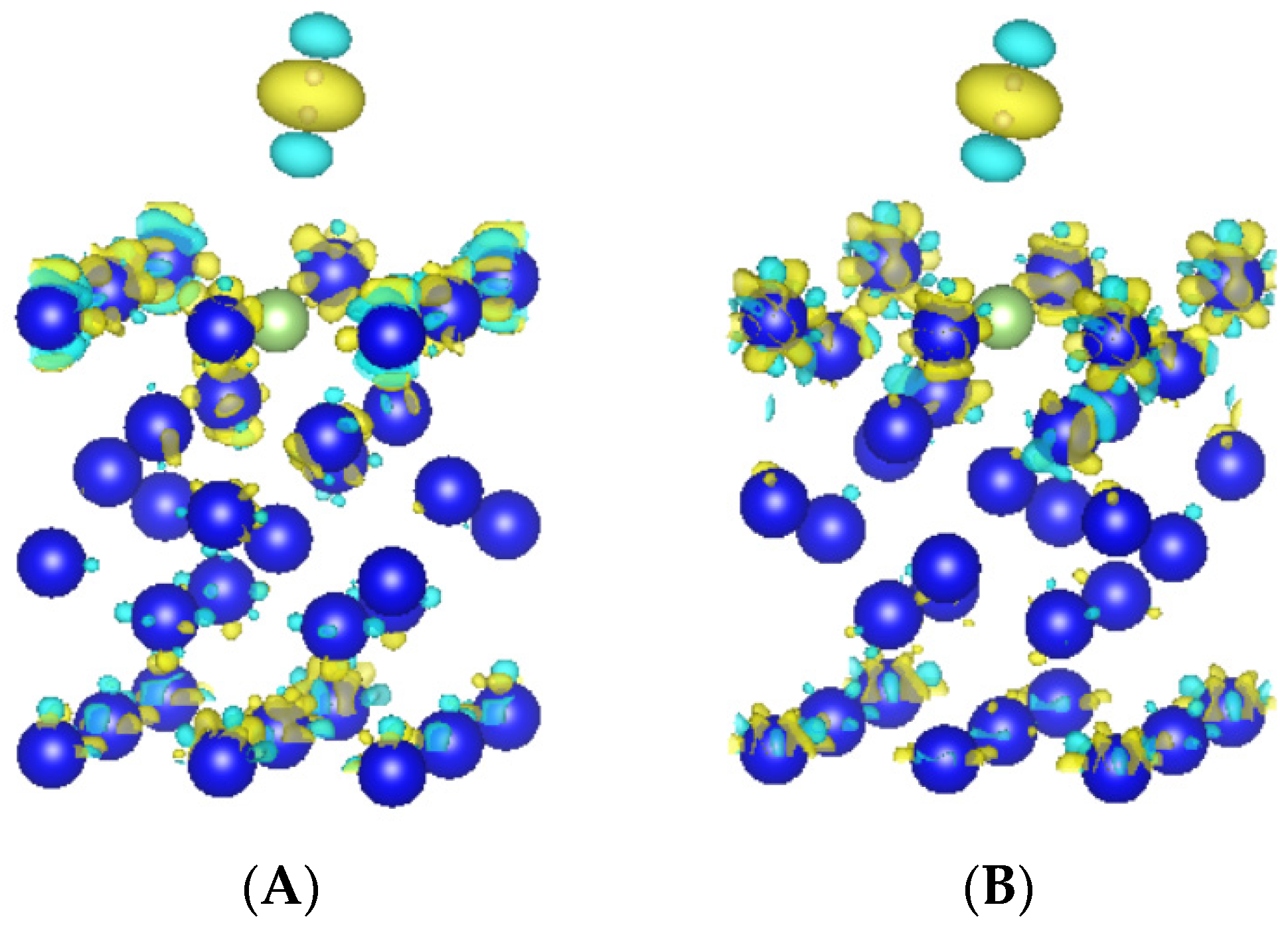
3.3.1. Differential Charge Density Analysis
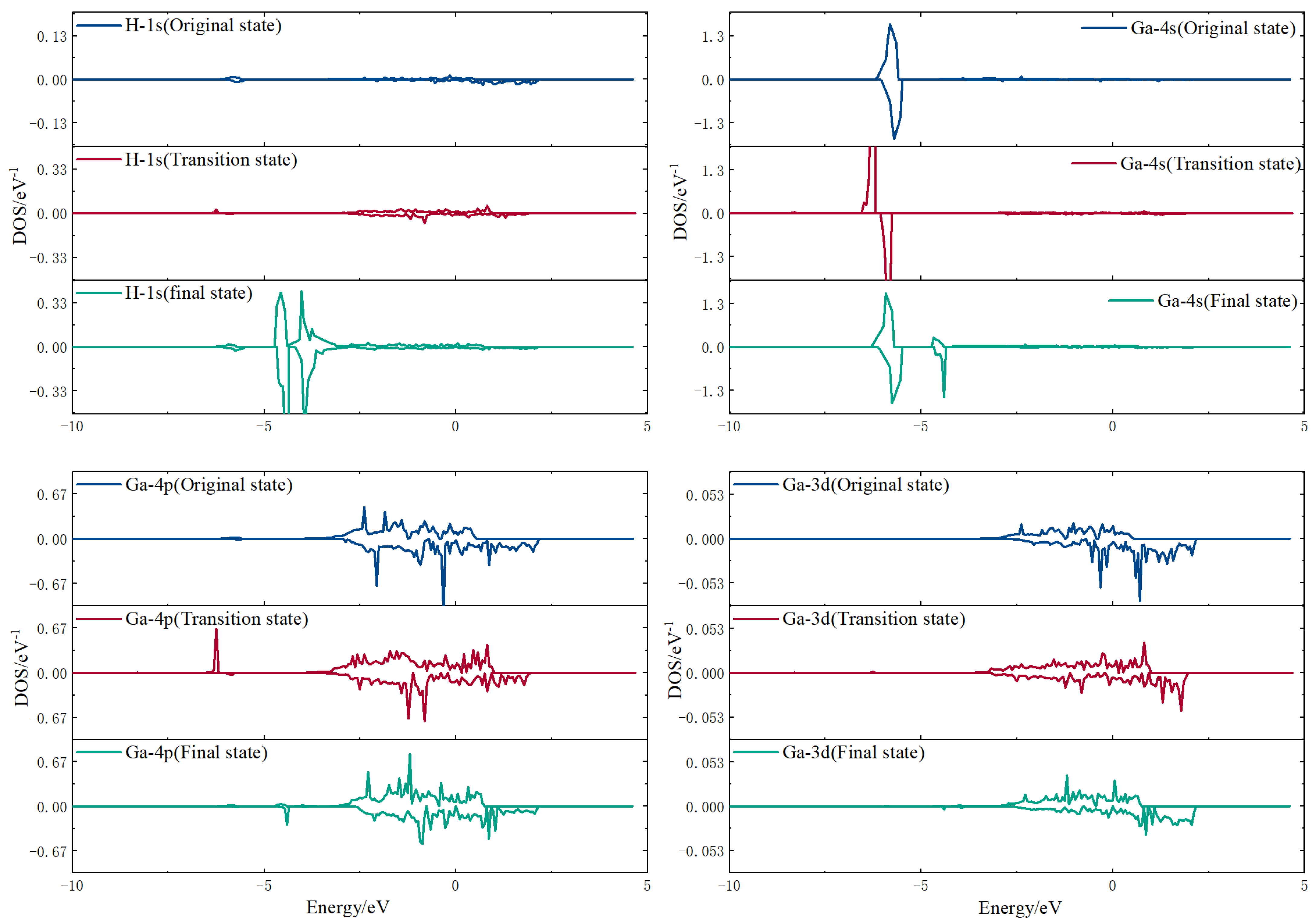
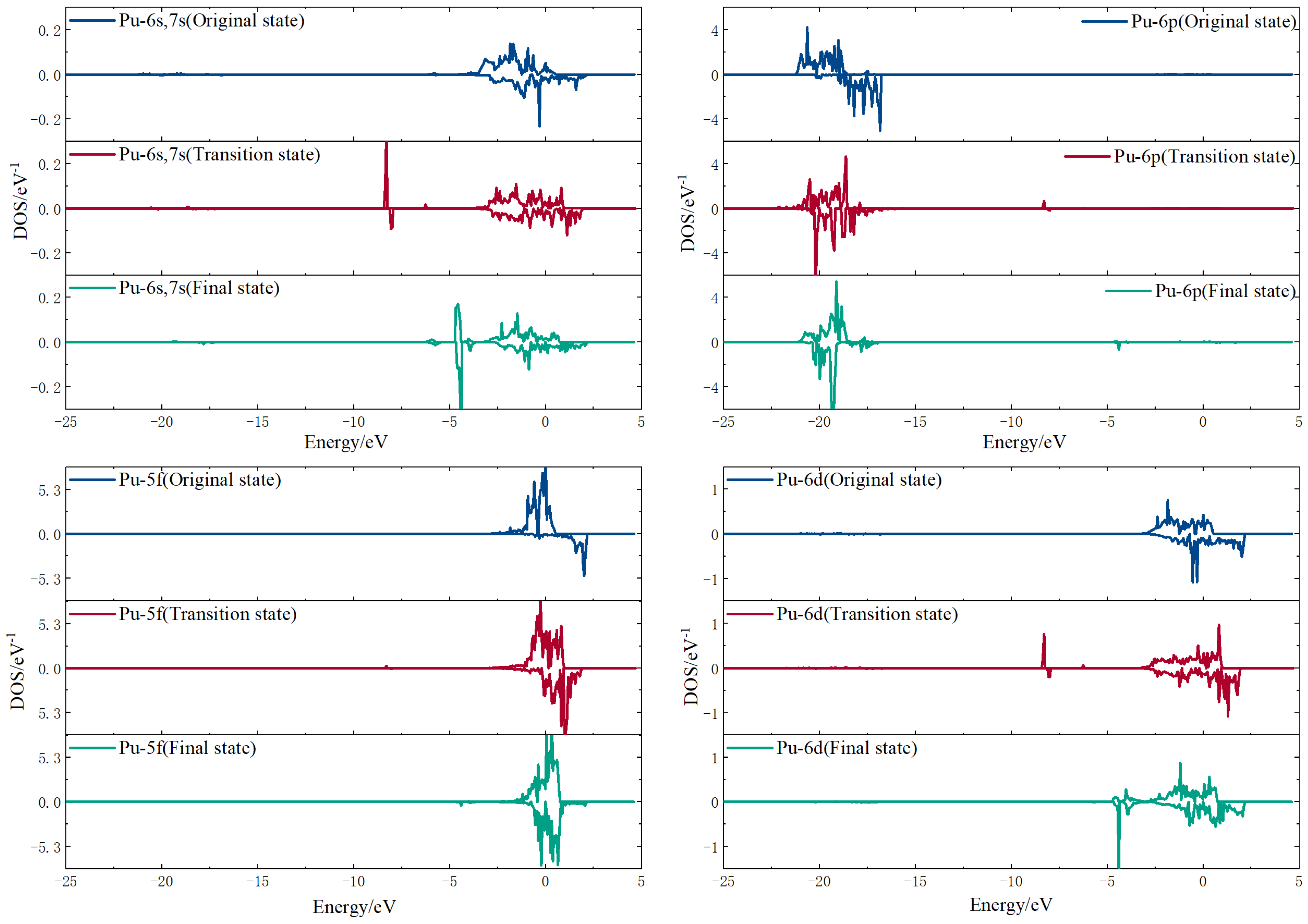
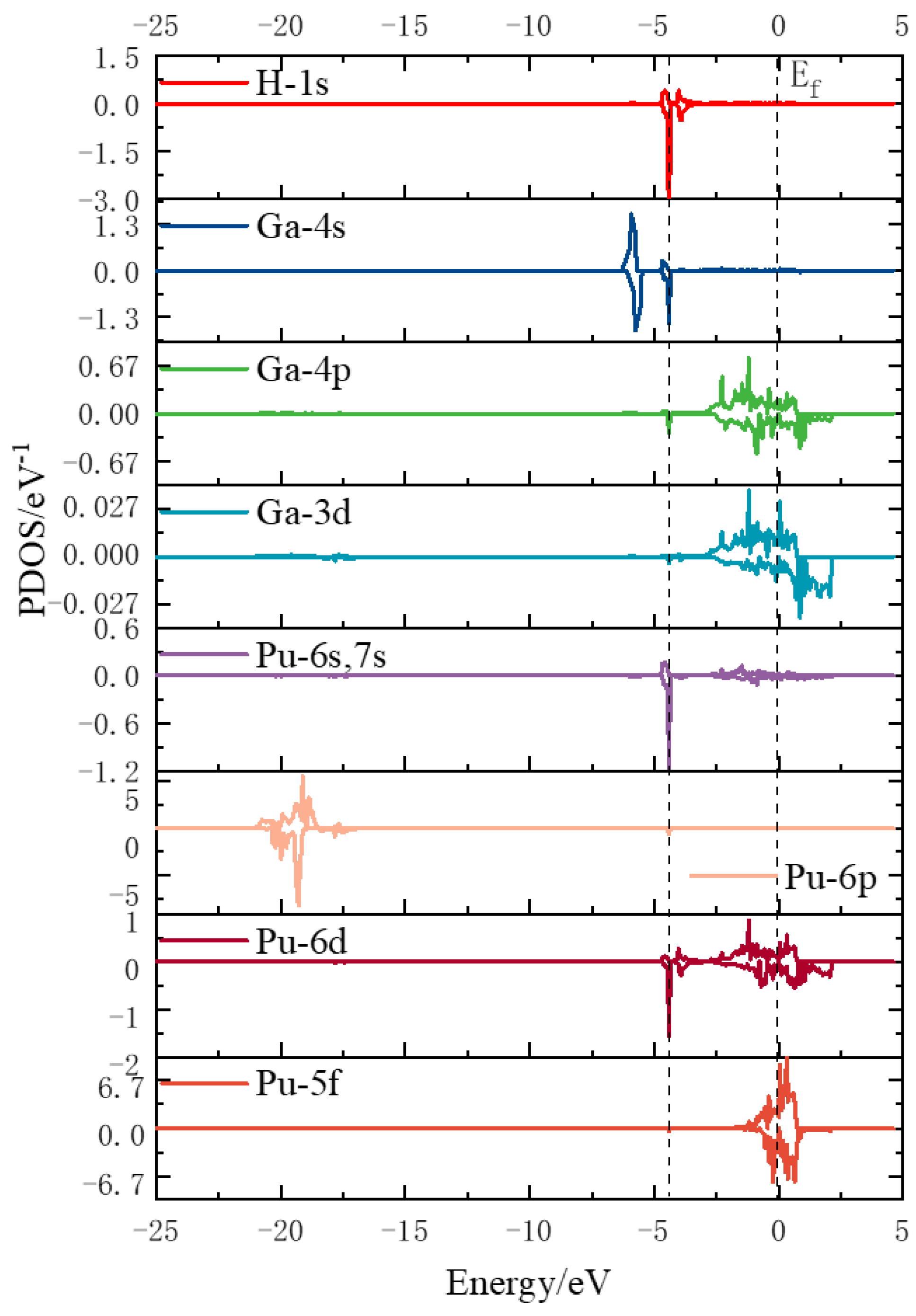
3.3.2. Electron Density of States Analysis
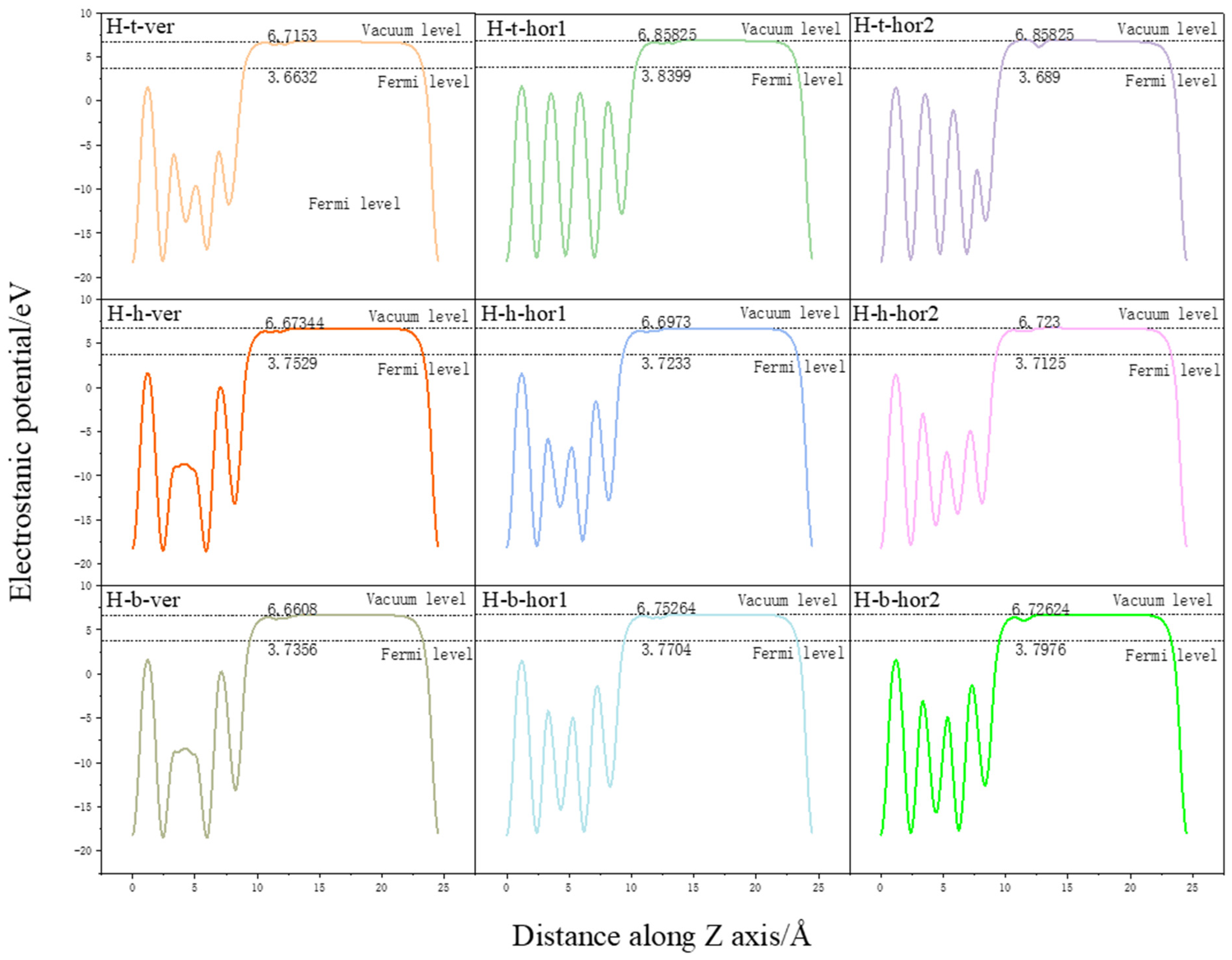
3.4. Surface Power Function Analysis
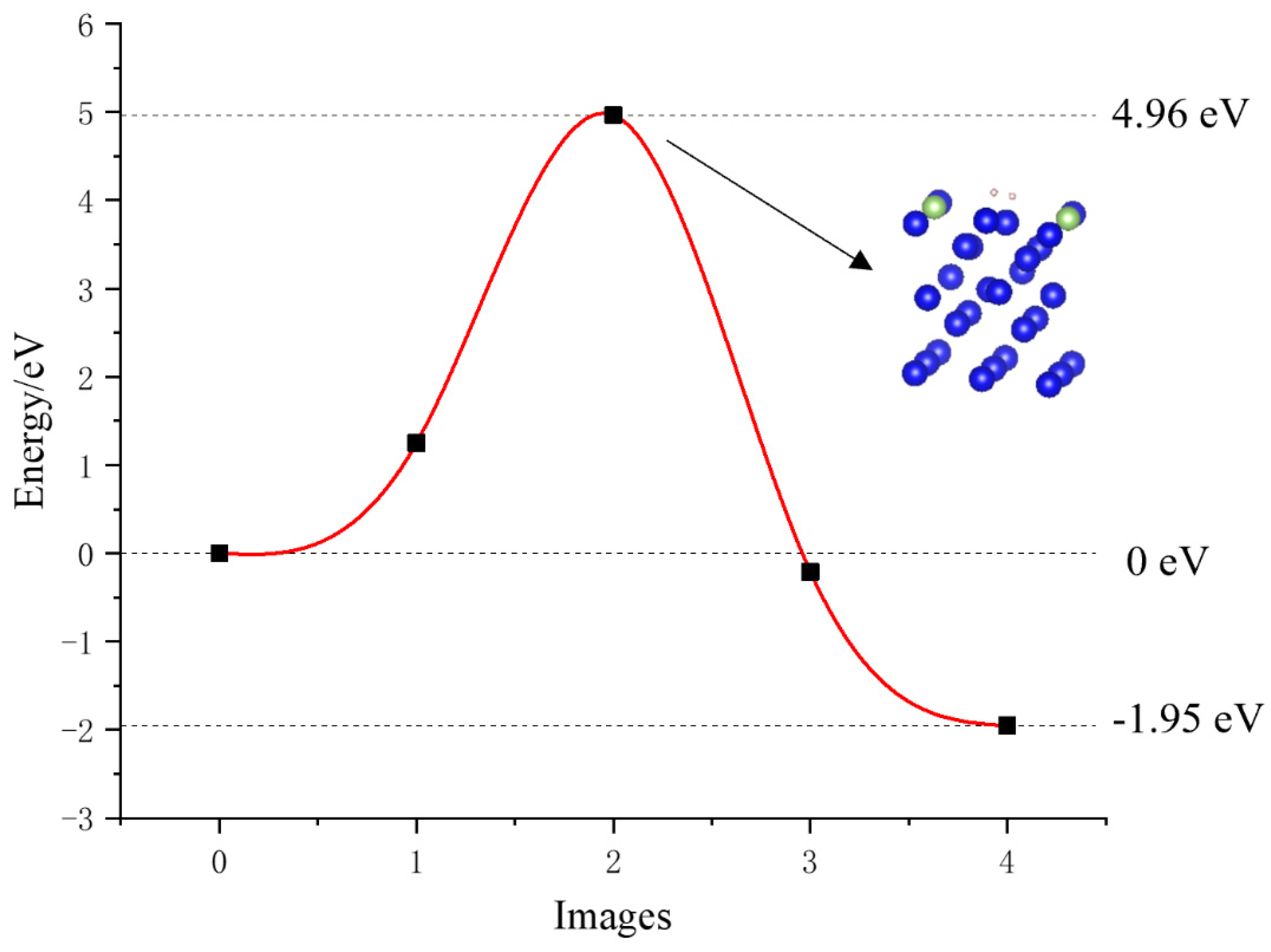
3.5. Dissociation of Hydrogen on the Surface of the Plutonium–Gallium System
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, G. Nuclear Fuel; Wu, S., Ed.; Chemical Industry Press: Beijing, China, 2007; p. 526. [Google Scholar]
- Hecker, S.S. Plutonium—An Element Never at Equilibrium. Metall. Mater. Trans. A 2008, 39, 1585–1592. [Google Scholar] [CrossRef]
- Shim, J.H.; Haule, K.; Kotliar, G. Fluctuating valence in a correlated solid and the anomalous properties of δ-plutonium. Nature 2007, 446, 513–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atta-Fynn, R.; Ray, A.K. A first principles study of the adsorption and dissociation of CO2 on the δ-Pu (111) surface. Eur. Phys. J. B Condens. Matter Complex Syst. 2009, 70, 171–184. [Google Scholar] [CrossRef]
- Goldman, N.; Morales, M.A. A First-Principles Study of Hydrogen Diffusivity and Dissociation on δ-Pu (100) and (111) Surfaces. J. Phys. Chem. C 2017, 121, 17950–17957. [Google Scholar] [CrossRef]
- Qi, C.B.; Wang, T.; Li, R.S.; Wang, J.T.; Qin, M.A.; Tao, S.H. First-principles study of co-adsorption behavior of O2 and CO2 molecules on δ-Pu(100) surface. Chin. Phys. B 2021, 30, 026601. [Google Scholar] [CrossRef]
- Cooper, N.G. Challenges in plutonium science. Los Alamos Sci. 2000, 26, 24–28. [Google Scholar]
- Hecker, S.S.; Martz, J.C. Aging of Plutonium and Its Alloys. Los Alamos Sci. 2000, 26, 238–243. [Google Scholar]
- Wick, O.J. Plutonium Handbook: A Guide to the Technology, 1st ed.; Gordon & Breach Science Publishers: Philadelphia, PA, USA, 1967; p. 2. [Google Scholar]
- Qi, C.; Wang, T.; Tao, S.; Qin, M. Research progress on surface corrosion of plutonium and its compounds. Rare Met. Mater. Eng. 2021, 6, 2008–2016. [Google Scholar]
- Sadigh, B.; Wolfer, W.G. Gallium stabilization of δ-Pu: Density-functional calculations. Phys. Rev. B 2005, 72, 205122. [Google Scholar] [CrossRef]
- Luo, W.; Meng, D.; Li, G.; Chen, H. Electronic Structure and Formation Heat of Pu3M and PuM3 (M = Ga, In, Sn and Ge) Compounds. Acta Phys.Chim. Sin. 2008, 24, 388–392. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.G.; Furthmüller, J.J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Jomard, G.; Bottin, F.; Geneste, G. Water adsorption and dissociation on the PuO2(110) surface. J. Nucl. Mater. 2014, 451, 28–34. [Google Scholar] [CrossRef]
- Hao, Y.G.; Eriksson, O.; Fernando, G.W.; Cooper, B.R. Surface electronic structure of gamma -uranium. Phys. Rev. B 1993, 47, 6680–6684. [Google Scholar] [CrossRef]
- Wei, H.; Hu, R.; Xiong, X.; Wang, G.; Song, H.; Luo, S. A first-principles study of H2 adsorption on δ-Pu(100) surface. J. Mol. Sci. 2010, 26, 5. [Google Scholar]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorption Conformation | Adsorption Energy Eads (eV) | Hydrogen Atomic Spacing RH-H (Å) | Hydrogen Atom to Surface Nearest Atom Distance | Nearest Distance of Hydrogen Atom to Surface | |||
|---|---|---|---|---|---|---|---|
| h(H1-Pu/Ga) (Å) | h(H2-Pu/Ga) (Å) | h(H1-Surface) (Å) | h(H2-Surface) (Å) | ||||
| H-t | ver | −0.5972 | 0.7536 | 3.518 | 4.2578 | 3.2807 | 4.0175 |
| hor1 | −0.3874 | 0.7541 | 3.4479 | 4.202 | 2.9886 | 3.7426 | |
| hor2 | −0.7016 | 0.7508 | 3.9653 | 4.0007 | 3.8741 | 3.9125 | |
| H-h | ver | −0.4985 | 0.7556 | 3.9795 | 4.4567 | 2.9704 | 3.7011 |
| hor1 | −1.4901 | 0.7546 | 3.8742 | 4.5208 | 2.9839 | 3.7277 | |
| hor2 | −0.9092 | 0.7537 | 3.6564 | 4.1832 | 3.0331 | 0.6979 | |
| H-b | ver | −0.3725 | 0.7522 | 3.8069 | 4.2528 | 3.4091 | 4.0641 |
| hor1 | −1.2954 | 0.7521 | 3.6248 | 4.3757 | 3.518 | 4.214 | |
| hor2 | −0.9014 | 0.7567 | 3.1307 | 3.287 | 2.8201 | 3.2091 | |
| Configuration | Adsorption of Atoms in Molecules | Total Number of Charges in Each Layer | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Q1 | Q2 | Qtotal | Q1st | Q2nd | Q3rd | Q4th | Q5th | ||
| H2 | 0.0148 | −0.0148 | 0 | - | - | - | - | - | |
| Exposed surfaces | - | - | - | −0.0049 | −0.0376 | 0.1573 | 0.1914 | −0.3061 | |
| H-t | ver | 0.0119 | 0.0074 | 0.0193 | −0.2804 | 0.2747 | 0.1492 | −0.0859 | −0.0772 |
| hor1 | 0.0420 | −0.0149 | 0.0271 | −0.2298 | −0.0220 | 0.0903 | 0.0496 | 0.0847 | |
| hor2 | 0.0203 | −0.0069 | 0.0133 | −0.3427 | 0.3256 | −0.0634 | −0.1204 | 0.1874 | |
| H-h | ver | 0.0382 | −0.0009 | 0.0372 | −0.1972 | −0.1812 | 0.6584 | −0.2145 | −0.1027 |
| hor1 | 0.0646 | −0.0286 | 0.0359 | −0.1688 | 0.0329 | 0.2225 | −0.046 | −0.0765 | |
| hor2 | 0.0286 | 0.0055 | 0.0341 | −0.2694 | 0.2883 | 0.0458 | −0.0822 | −0.0169 | |
| H-b | ver | 0.0135 | 0.0117 | 0.0252 | −0.1566 | −0.1653 | 0.7305 | −0.2923 | −0.1416 |
| hor1 | 0.0124 | 0.0113 | 0.0238 | −0.1018 | 0.0201 | 0.1106 | −0.0043 | −0.0485 | |
| hor2 | 0.0285 | 0.0061 | 0.0346 | −0.1404 | 0.1259 | 0.0261 | −0.0533 | 0.0068 | |
| Adsorption Conformation | Vacuum Energy Level/eV | Fermi Energy Level/eV | Power Letter/eV | ΔΦ/eV |
|---|---|---|---|---|
| H-t-ver | 6.7153 | 3.6632 | 3.0521 | 0.0626 |
| H-t-hor1 | 6.8662 | 3.8399 | 3.0263 | 0.0368 |
| H-t-hor2 | 6.8689 | 3.6890 | 3.0799 | 0.0903 |
| H-h-ver | 6.6836 | 3.7529 | 2.9307 | −0.0588 |
| H-h-hor1 | 6.6973 | 3.7233 | 2.9740 | −0.0155 |
| H-h-hor2 | 6.7230 | 3.7125 | 3.0105 | 0.0210 |
| H-b-ver | 6.6608 | 3.7356 | 2.9252 | −0.0642 |
| H-b-hor1 | 6.7478 | 3.7704 | 2.9774 | −0.0120 |
| H-b-hor2 | 6.7138 | 3.7976 | 2.9162 | −0.0733 |
| Pure Surface | 6.7816 | 3.7921 | 2.9895 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Zhu, M.; Huang, H.; Liang, T.; Su, H.; Xue, B. First-Principles Study on the Adsorption and Dissociation Behavior of H2 on the Surface of a Plutonium–Gallium System. Coatings 2022, 12, 1019. https://doi.org/10.3390/coatings12071019
Li L, Zhu M, Huang H, Liang T, Su H, Xue B. First-Principles Study on the Adsorption and Dissociation Behavior of H2 on the Surface of a Plutonium–Gallium System. Coatings. 2022; 12(7):1019. https://doi.org/10.3390/coatings12071019
Chicago/Turabian StyleLi, Longxian, Min Zhu, Huang Huang, Tianxi Liang, Huan Su, and Baiyu Xue. 2022. "First-Principles Study on the Adsorption and Dissociation Behavior of H2 on the Surface of a Plutonium–Gallium System" Coatings 12, no. 7: 1019. https://doi.org/10.3390/coatings12071019