
Electronic Structure of Graphene on the Hexagonal Boron Nitride Surface: A Density Functional Theory Study


Abstract
:1. Introduction
2. Structures and Methods
3. Results
3.1. Structural Parameters
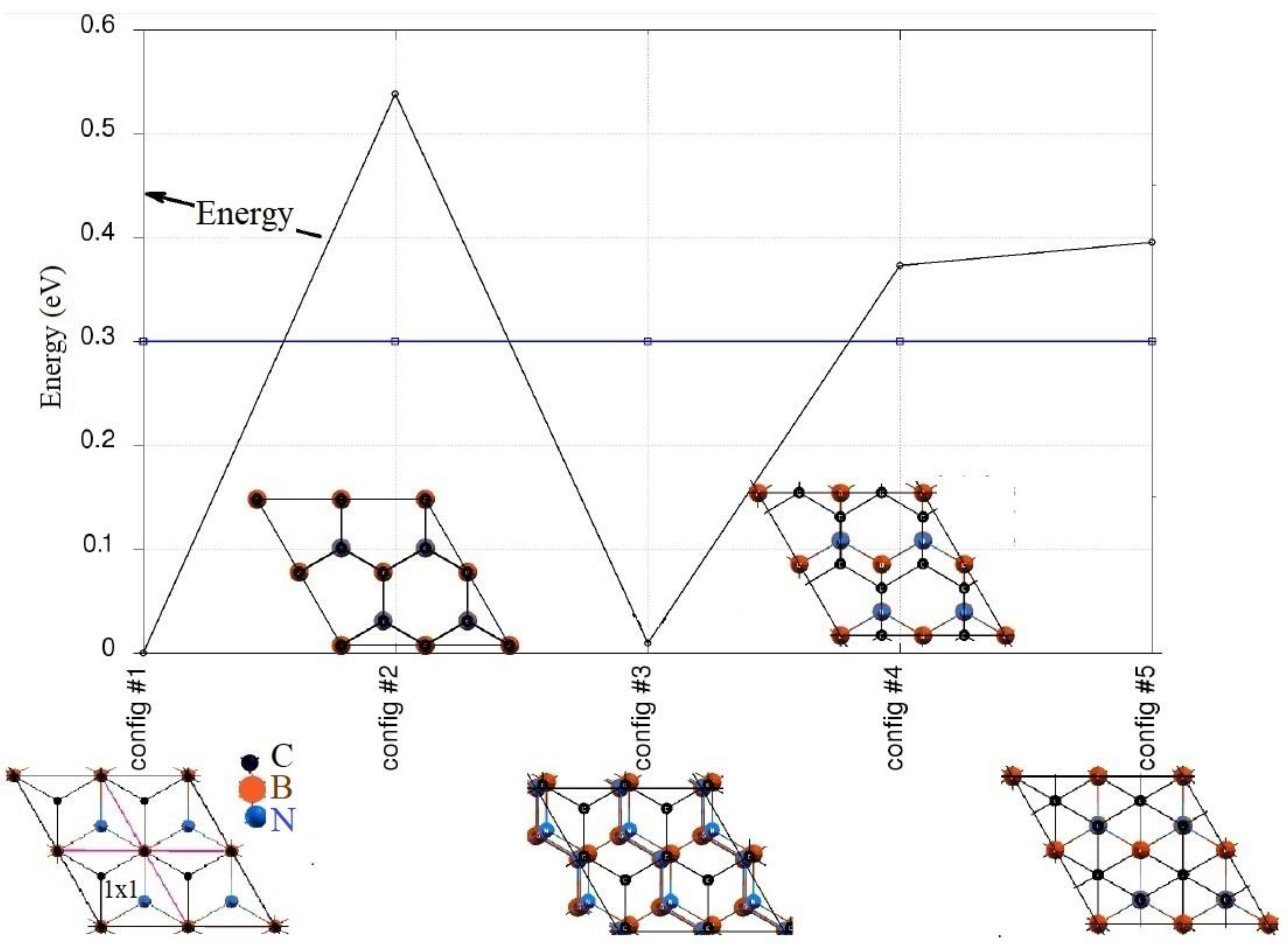
3.1.1. Structural Properties of the A-Lattice
3.1.2. Structural Properties of the B-Lattice
3.2. Electronic Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and graphene oxide: Synthesis, properties, and applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef] [PubMed]
- Avouris, P.; Dimitrakopoulos, C. Graphene: Synthesis and applications. Mater. Today 2012, 15, 86–97. [Google Scholar] [CrossRef]
- Soldano, C.; Mahmood, A.; Dujardin, E. Production, properties and potential of graphene. Carbon 2010, 48, 2127–2150. [Google Scholar] [CrossRef] [Green Version]
- Batzil, M. The surface science of graphene: Metal interfaces, CVD synthesis, nanoribbons, chemical modifications, and defects. Surf. Sci. Rep. 2012, 67, 83–115. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Singha, V.; Joung, D.; Zhai, L.; Das, D.; Khondaker, S.I.; Seal, S. Graphene based materials: Past, present and future. Prog. Mater. Sci. 2011, 56, 1178–1271. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Morozov, S.V.; Mohinddin, T.M.G.; Ponomarenko, L.A.; Elias, D.C.; Yang, R.; Barbolina, I.I.; Blake, P.; Booth, T.J.; Jiang, D.; et al. Electronic properties of graphene. Phys. Stat. Sol. B 2007, 244, 4106–4111. [Google Scholar] [CrossRef] [Green Version]
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of grapheme. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.-M.; Jhi, S.-H.; Son, Y.-W. Effects of strain on electronic properties of graphene. Phys. Rev. B 2010, 81, 081407. [Google Scholar] [CrossRef] [Green Version]
- Abergel, D.S.L.; Apalkov, V.; Berashevich, J.; Ziegler, K.; Chakraborty, T. Properties of graphene: A theoretical perspective. Adv. Phys. 2010, 59, 261–482. [Google Scholar] [CrossRef] [Green Version]
- Rico, M.J.E.; Rodríguez-Martínez, J.A.; Moreno-Armenta, M.G.; Takeuchi, N. Graphene monolayers on GaN(0001). Appl. Surf. Sci. 2015, 326, 7–11. [Google Scholar] [CrossRef]
- Frank, I.W.; Tanenbaum, D.M.; van der Zande, A.M.; McEuen, P.L. Mechanical properties of suspended graphene sheets. J. Vac. Sci. Technol. B Microelectron. Nanometer Struct. 2007, 25, 2558. [Google Scholar] [CrossRef] [Green Version]
- Min, K.; Aluru, N.R. Mechanical properties of graphene under shear deformation. Appl. Phys. Lett. 2011, 98, 013113. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.; Tsai, I.-L.; Sepioni, M.; Lehtinen, O.; Keinonen, J.; Krasheninnikov, A.; Neto, A.C.; Katsnelson, M.; Geim, A.; Grigorieva, I. Dual origin of defect magnetism in graphene and its reversible switching by molecular doping. Nat. Commun. 2013, 4, 2010. [Google Scholar] [CrossRef] [PubMed]
- Taychatanapat, T.; Wang, H.; Hsu, A.; Watanabe, K.; Taniguchi, T.; Jarillo-Herrero, P.; Palacio, T. BN/graphene/BN transistors for RF applications. IEEE Electron. Device Lett. 2011, 32, 1209–1211. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.A.; McGlynn, A.G.; Towlson, B.M.; Gunn, M.; Jones, D.; Jenkins, T.E.; Winter, R.; Poolton, N.R.J. Determination of the optical band-gap energy of cubic and hexagonal boron nitride using luminescence excitation spectroscopy. J. Phys. Condens. Matter. 2008, 20, 075233. [Google Scholar] [CrossRef]
- Pittroff, W.; Erbert, G.; Beister, G.; Bugge, F.; Klein, F.A.; Knauer, A.; Maege, J.; Ressel, P.; Sebastian, J.; Staske, R.; et al. Mounting of high power laser diodes on boron nitride heat sinks using an optimized Au/Sn metallurgy. IEEE Trans. Adv. 2001, 24, 434–441. [Google Scholar] [CrossRef]
- Subramani, S.; Devarajan, M. Thermal transient analysis of high-power green LED fixed on BN coated Al substrates as heatsink. IEEE Trans. Electron Devices 2014, 61, 3213–3216. [Google Scholar] [CrossRef]
- Takahashia, S.; AkinoriKana, Y.; Hottab, Y.; Ogawa, H. Dielectric and thermal properties of isotactic polypropylene/hexagonal boron nitride composites for high-frequency applications. J. Alloys Compd. 2014, 615, 141–145. [Google Scholar] [CrossRef]
- Tobar, Á.H.; López, C.O.; Martínez, J.A.R. Study of the structural and electronic properties of three- and two-dimensional transition-metal dioxides using first-principles calculations. Comput. Condens. Matter. 2020, 25, e00498. [Google Scholar] [CrossRef]
- Watanabea, K.; Taniguchia, T.; Miyab, K.; Satob, Y.; Nakamurab, K.; Niiyamab, T.; Taniguchi, M. Hexagonal boron nitride as a new ultraviolet luminescent material and its application—Fluorescence properties of hBN single-crystal powder. Diam. Relat. Mater. 2011, 20, 849–852. [Google Scholar] [CrossRef]
- Ishida, H.; Rimdusit, S. Very high thermal conductivity obtained by boron nitride-filled polybenzoxazine. Thermochim. Acta 1998, 320, 177–186. [Google Scholar] [CrossRef]
- Xu, Y.-N.; Ching, W.Y. Calculation of ground-state and optical properties of boron nitrides in the hexagonal, cubic, and wurtzite structures. Phys. Rev. B 1991, 44, 7787–7798. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, Y.P.; Shen, Z.X. Structural and electronic properties of h-BN. Phys. Rev. B. 2003, 68, 104102. [Google Scholar] [CrossRef]
- Wang, J.; Ma, F.; Liang, W.; Sun, M. Electrical properties and applications of graphene, hexagonal boron nitride (h-BN), and graphene/h-BN heterostructures. Mater. Today Phys. 2017, 2, 6–34. [Google Scholar] [CrossRef]
- Gilje, S.; Han, S.; Wang, M.; Wang, K.L.; Kaner, R.B. A chemical route to graphene for device applications. Nano Lett. 2007, 7, 3394–3398. [Google Scholar] [CrossRef]
- Nam, Y.; Lindvall, N.; Sun, J.; Park, Y.W.; Yurgens, A. Graphene p–n–p junctions controlled by local gates made of naturally oxidized thin aluminium films. Carbon 2012, 50, 1987–1992. [Google Scholar] [CrossRef]
- Li, X.; Cai, W.; An, J.; Kim, S.; Nah, J.; Yang, D.; Piner, R.; Velamakanni, A.; Jung, I.; Tutuc, E.; et al. Large-area synthesis of high-quality and uniform graphene films on copper foils. Science 2009, 324, 1312–1314. [Google Scholar] [CrossRef] [Green Version]
- Giovannetti, G.; Khomyakov, P.A.; Brocks, G.; Kelly, P.J.; Brink, J. Substrate-induced band gap in graphene on hexagonal boron nitride: Ab initio density functional calculations. Phys. Rev. B 2007, 76, 073103. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Laasonen, K.; Pasquarello, A.; Car, R.; Lee, C.; Vanderbilt, D. Car-Parrinello molecular dynamics with Vanderbilt ultrasoft pseudopotentials. Phys. Rev. B 1993, 47, 10142–10153. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, 864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, 1133–1138. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.; Sa, B.; Zhou, J.; Ahuja, R.; Sun, Z. Design of High-efficiency visible-light photocatalysts for water splitting: MoS2/AlN(GaN) heterostructures. J. Phys. Chem. C 2014, 118, 17594–17599. [Google Scholar] [CrossRef]
- Fan, Y.; Ma, X.; Liu, X.; Wang, J.; Ai, H.; Zhao, M. Theoretical design of an InSe/GaTe vdW heterobilayer: A potential visible-light photocatalyst for water splitting. J. Phys. Chem. C 2018, 122, 27803–27810. [Google Scholar] [CrossRef]
- Ramasubramaniam, A.; Naveh, D.; Towe, E. Tunable band gaps in bilayer graphene−BN heterostructures. Nano Lett. 2011, 11, 1070–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Zhao, M.; Wang, Z.; Zhang, X.; Zhang, H. Tunable electronic structures of graphene/boron nitride heterobilayers. Appl. Phys. Lett. 2011, 98, 083103. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, Y.-K.; Kim, H.-J.; Scheicher, R.H.; Cho, J.-H. Physisorption of DNA Nucleobases on h-BN and Graphene: vdW-Corrected DFT Calculations. J. Phys. Chem. C 2013, 117, 13435–13441. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Ertekin, E. Phonon transport on two-dimensional graphene/boron nitride superlattices. Phys. Rev. B 2014, 90, 195209. [Google Scholar] [CrossRef]
- Özçelik, V.O.; Ciraci, S. Nanoscale dielectric capacitors composed of graphene and boron nitride layers: A first-principles study of high capacitance at nanoscale. J. Phys. Chem. C 2013, 117, 15327–15334. [Google Scholar] [CrossRef] [Green Version]
- Sachs, B.; Wehling, T.O.; Katsnelson, M.I.; Lichtenstein, A.I. Adhesion and electronic structure of graphene on hexagonal boron nitride substrates. Phys. Rev. B 2011, 84, 195414. [Google Scholar] [CrossRef] [Green Version]
- Bawari, S.; Kaley, N.M.; Pal, S.; Vineesh, T.V.; Ghosh, S.; Mondal, J.; Narayanan, T.N. On the hydrogen evolution reaction activity of graphene–hBN van der Waals heterostructures. Phys. Chem. Chem. Phys. 2018, 20, 15007–15014. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Jiang, D.; Schedin, F.; Booth, T.J.; Khotkevich, V.V.; Morozov, S.V.; Geim, A.K. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. USA 2005, 102, 10451–10453. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Taniguchi, T.; Kanda, H. Direct-bandgap properties and evidence for ultraviolet lasing of hexagonal boron nitride single crystal. Nat. Mater. 2004, 3, 404–409. [Google Scholar] [CrossRef]
- Gorbachev, R.V.; Riaz, I.; Nair, R.R.; Jalil, R.; Britnell, L.; Belle, B.D.; Hill, E.W.; Novoselov, K.S.; Watanabe, K.; Taniguchi, T.; et al. Hunting for monolayer boron nitride: Optical and Raman signatures. Small 2011, 7, 465–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Q.; Lv, R. Two-dimensional heterostructures based on graphene and transition metal dichalcogenides: Synthesis, transfer and applications. Carbon 2019, 145, 240–250. [Google Scholar] [CrossRef]
- Hu, W.; Yang, Y. First-principles study of two-dimensional van der Waals heterojunctions. Comput. Mater. Sci. 2019, 112, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Yankowitz, M.; Ma, Q.; Jarillo-Herrero, P.; Leroy, B.J. van der Waals heterostructures combining graphene and hexagonal boron nitride. Nat. Rev. Phys. 2019, 1, 112–125. [Google Scholar] [CrossRef]
- Peyghan, A.A.; Noei, M.; Yourdkhani, S. Al-doped graphene-like BN nanosheet as a sensor for para-nitrophenol: DFT study. Superlattices Microstruct. 2013, 59, 115–122. [Google Scholar] [CrossRef]
- Behera, H.; Mukhopadhyay, G. Strain-tunable bandgap in graphene/h-BN hetero-bilayer. J. Phys. Chem. Solids 2012, 73, 818–821. [Google Scholar] [CrossRef] [Green Version]
- Lui, C.H.; Li, Z.; Mak, K.F.; Cappelluti, E.; Heinz, T.F. Observation of an electrically tunable band gap in trilayer graphene. Nat. Phys. 2011, 7, 944–947. [Google Scholar] [CrossRef] [Green Version]
- Kharche, N.; Nayak, S.K. Quasiparticle band gap engineering of graphene and graphone on hexagonal boron nitride substrate. Nano Lett. 2011, 11, 5274–5278. [Google Scholar] [CrossRef] [Green Version]
- Sevilla, J.R.; Putungan, D.B. Graphene-hexagonal boron nitride van der Waals heterostructures: An examination of the effects of different van der Waals corrections. Mater. Res. Express 2021, 8, 085601. [Google Scholar] [CrossRef]
- Chen, Z.-G.; Shi, Z.; Yang, W.; Lu, X.; Lai, Y.; Yan, H.; Wang, F.; Zhang, G.; Li, Z. Observation of an intrinsic bandgap and Landau level renormalization in graphene/boron-nitride heterostructures. Nat. Commun. 2014, 5, 4461. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; DaSilva, A.M.; MacDonald, A.H.; Adam, S. Origin of band gaps in graphene on hexagonal boron nitride. Nat. Commun. 2015, 6, 6308. [Google Scholar] [CrossRef] [Green Version]
- Bokdam, M.; Amlaki, T.; Brocks, G.; Kelly, P.J. Band gaps in incommensurable graphene on hexagonal boron nitride. Phys. Rev. B 2014, 89, 201404. [Google Scholar] [CrossRef] [Green Version]
- Hunt, B.; Taniguchi, T.; Moon, P.; Koshino, M.; Ashoori, R.C. Massive dirac fermions and hofstadter butterfly in a van der Waals heterostructure. Science 2013, 340, 1427–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structures | aBN (Å) | lB-N (Å) | agraphene (Å) | lC-C (Å) | Kinetic Energy Cutoff | Charge Energy Cutoff | K-Points |
|---|---|---|---|---|---|---|---|
| A-lattice | 2.510 | 1.449 | 2.444 | 1.411 | 45 | 450 | 6 × 6 × 3 |
| B-lattice | 2.510 | 1.449 | 2.444 | 1.411 | 35 | 350 | 6 × 6 × 3 |
| Structure | Calculated | |
|---|---|---|
| A-lattice | In this paper | −100.300 |
| B-lattice | In this paper | −89.800 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casiano-Jiménez, G.; Ortega-López, C.; Rodríguez-Martínez, J.A.; Moreno-Armenta, M.G.; Espitia-Rico, M.J. Electronic Structure of Graphene on the Hexagonal Boron Nitride Surface: A Density Functional Theory Study. Coatings 2022, 12, 237. https://doi.org/10.3390/coatings12020237
Casiano-Jiménez G, Ortega-López C, Rodríguez-Martínez JA, Moreno-Armenta MG, Espitia-Rico MJ. Electronic Structure of Graphene on the Hexagonal Boron Nitride Surface: A Density Functional Theory Study. Coatings. 2022; 12(2):237. https://doi.org/10.3390/coatings12020237
Chicago/Turabian StyleCasiano-Jiménez, Gladys, César Ortega-López, Jairo Arbey Rodríguez-Martínez, María Guadalupe Moreno-Armenta, and Miguel J. Espitia-Rico. 2022. "Electronic Structure of Graphene on the Hexagonal Boron Nitride Surface: A Density Functional Theory Study" Coatings 12, no. 2: 237. https://doi.org/10.3390/coatings12020237