Broad-Spectrum Antimicrobial Activity and Improved Stability of a D-Amino Acid Enantiomer of DMPC-10A, the Designed Derivative of Dermaseptin Truncates

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
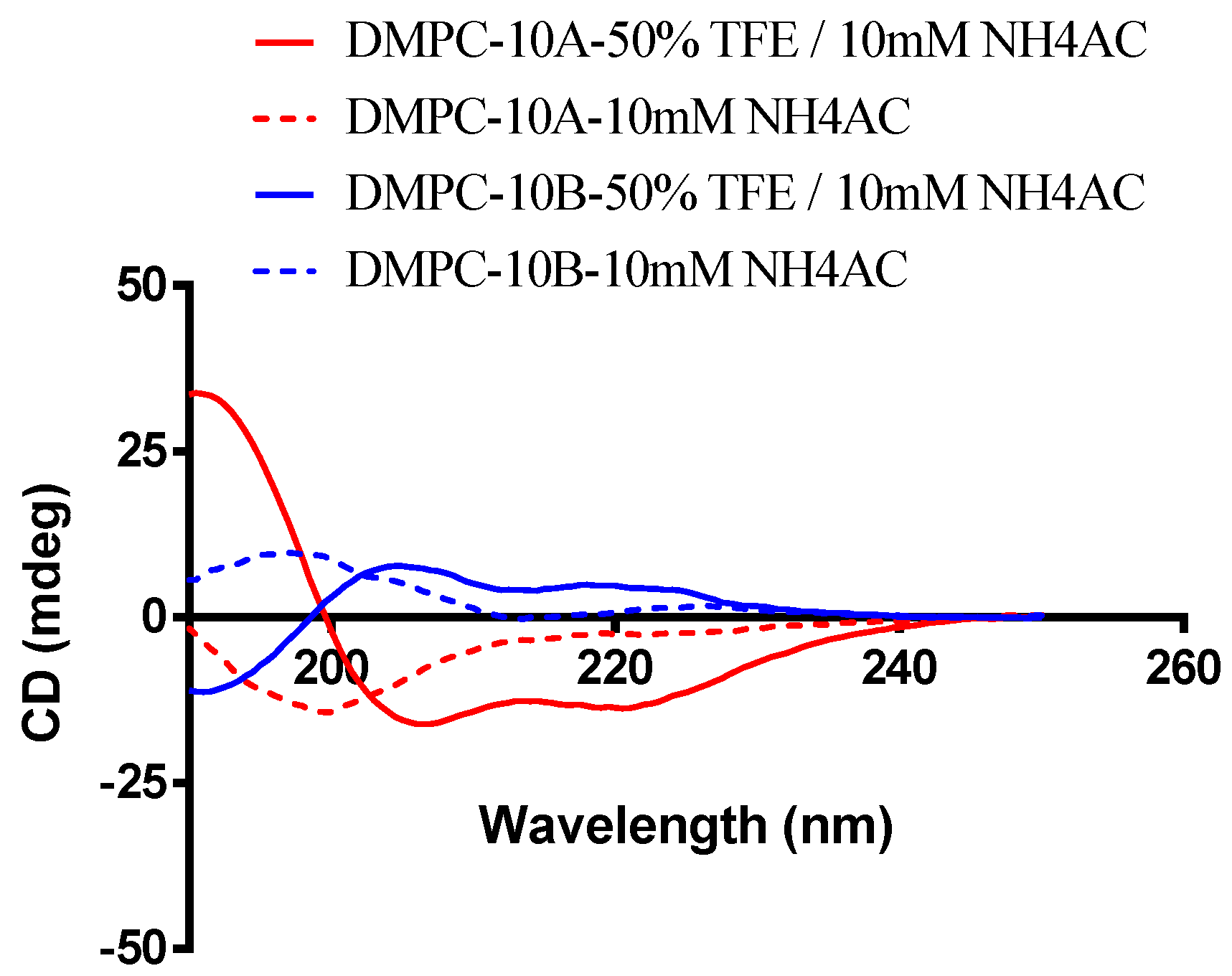
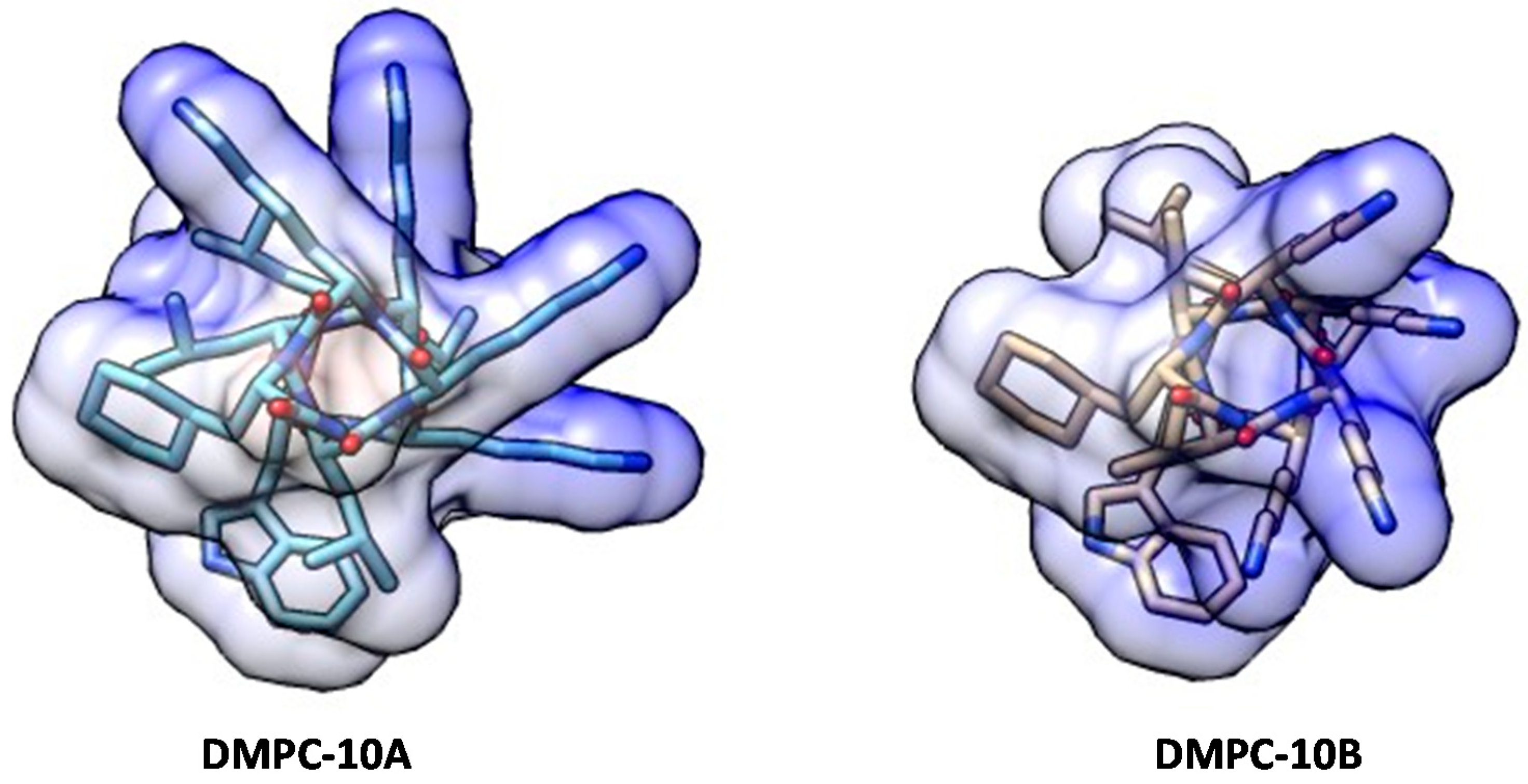
2.1. Structure Analysis of DMPC-10B
2.2. Antimicrobial Activities
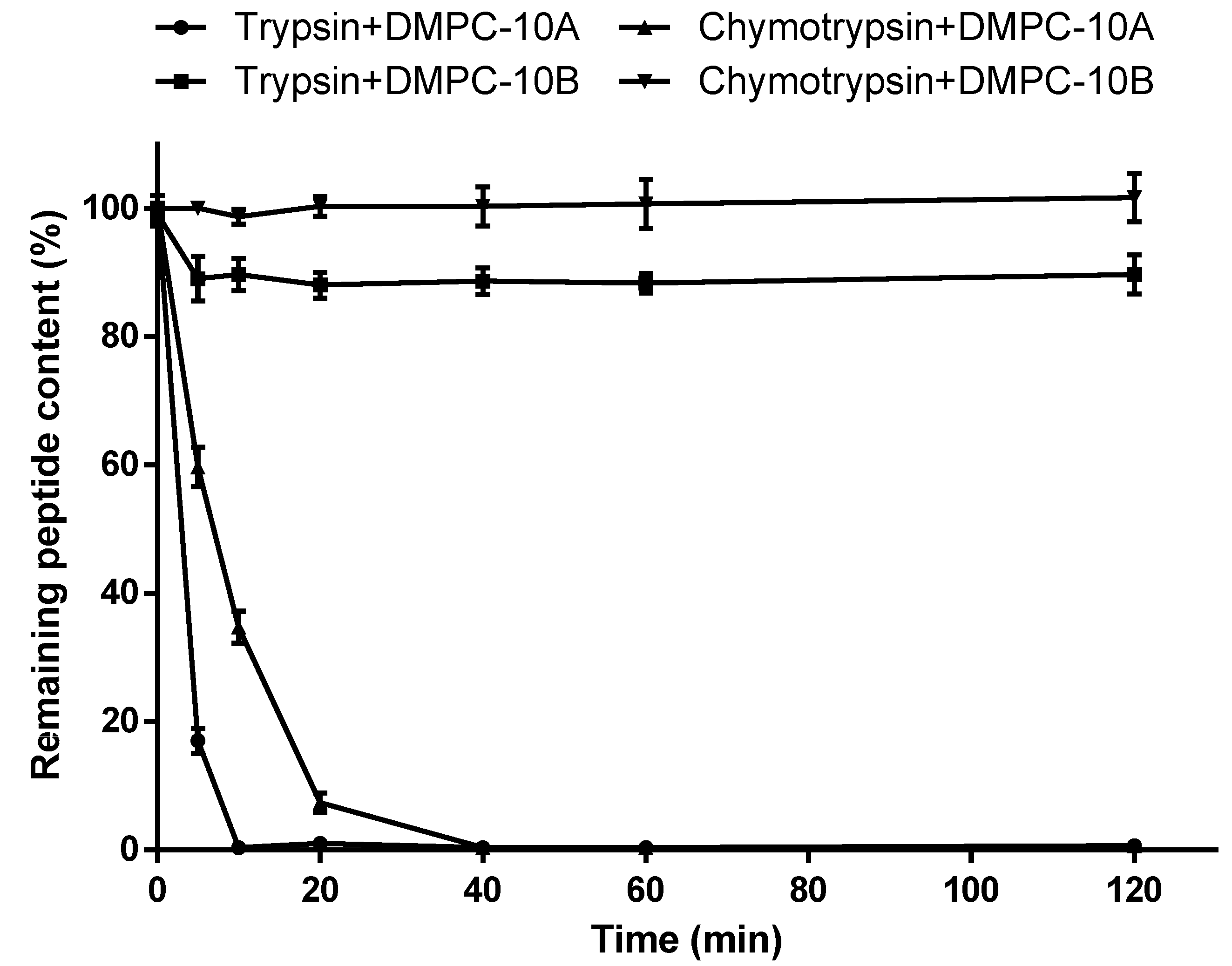
2.3. Enzyme Stability
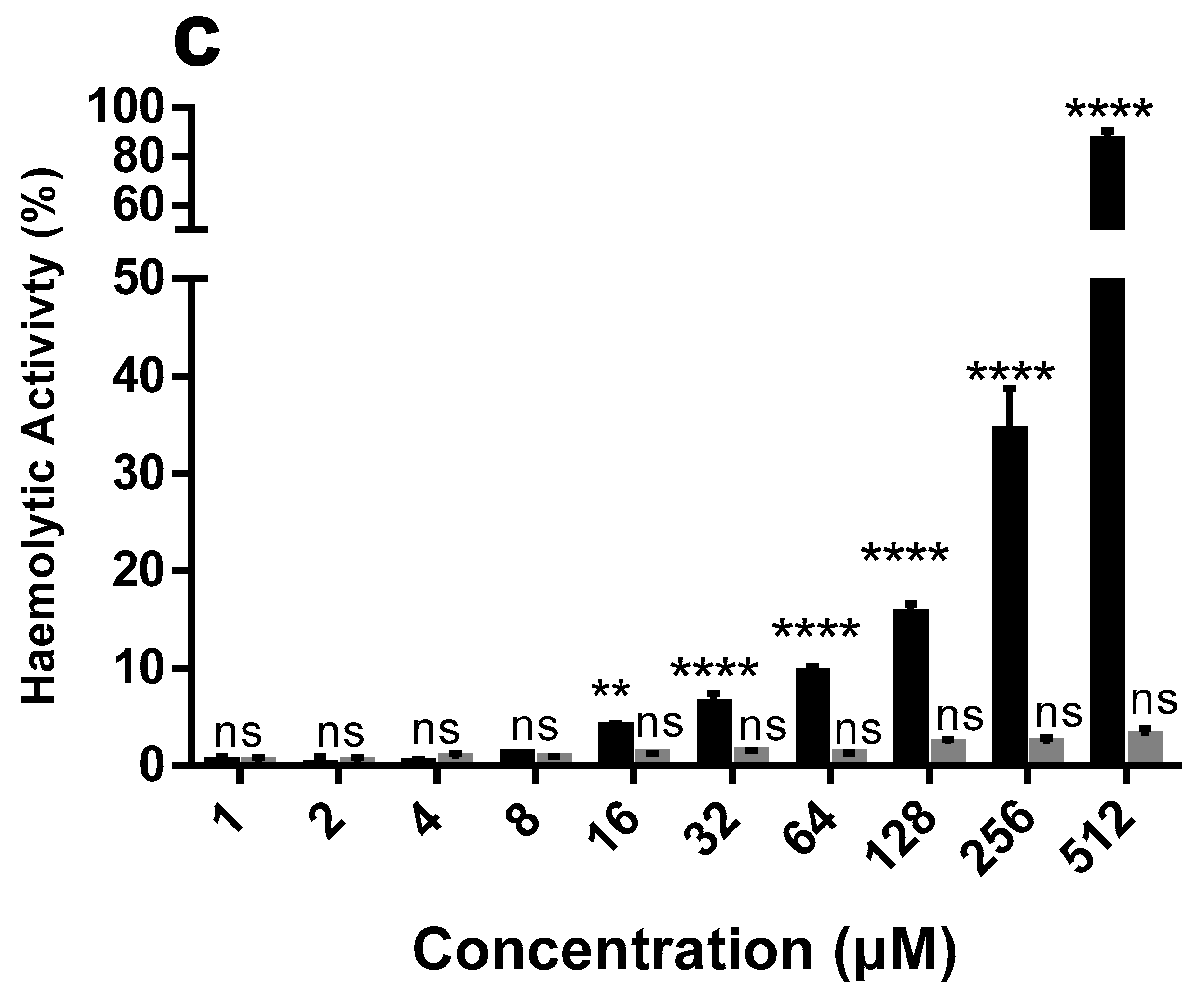
2.4. Hemolytic and Cytotoxic Activity
2.5. Membrane Permeability
2.6. Antibiofilm Activities
2.7. Antimicrobial Synergy Study
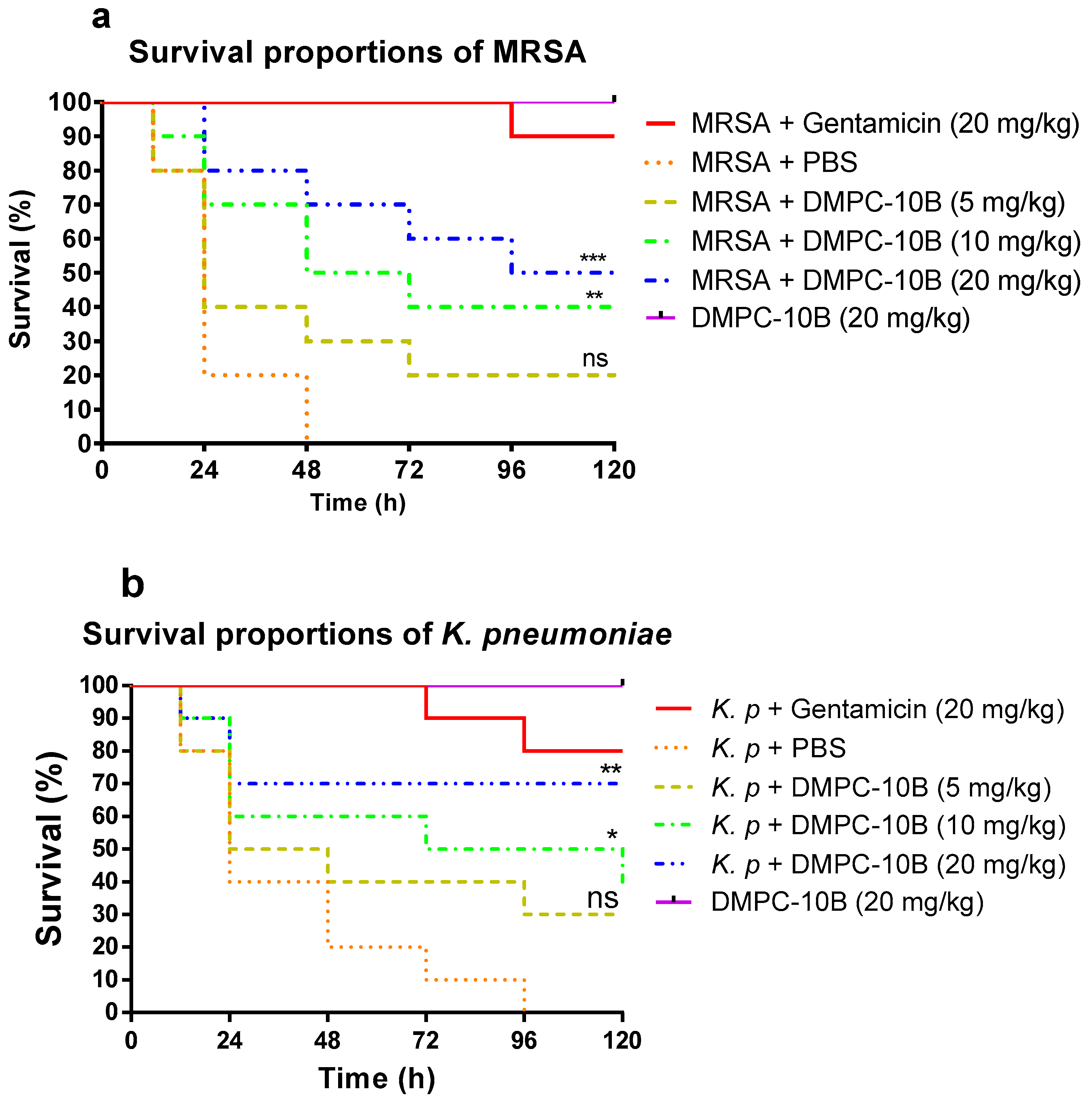
2.8. Treatment of Larvae Infected with MRSA and K. pneumoniae with DMPC-10B
3. Discussion
4. Materials and Methods
4.1. Solid Phase Peptide Synthesis
4.2. Molecular Modeling and Docking
4.3. Circular Dichroism (CD)
4.4. Antimicrobial Assay
4.5. Enzyme Stability Assay
4.6. Cytotoxicity Assay
4.7. Hemolysis Test
4.8. Membrane Permeability Kinetic Assay
4.9. Antibiofilm Assays
4.10. Evaluation of Combination Effects of DMPC-10B
4.11. Assessing the Efficacy of DMPC-10B against MRSA and K. Pneumoniae Strains In Vivo
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cordes, J.; Wittersheim, M.; Harder, J.; Glaser, R. The skin’s own antibiotics. Important features of antimicrobial peptides for clinical practice. Hautarzt 2014, 65, 50–55. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Hein-Kristensen, L.; Franzyk, H.; Holch, A.; Gram, L. Adaptive Evolution of Escherichia coli to an alpha-Peptide/beta-Peptoid Peptidomimetic Induces Stable Resistance. PLoS ONE 2013, 8, e73620. [Google Scholar] [CrossRef] [Green Version]
- Ravensdale, J.; Wong, Z.; O’Brien, F.; Gregg, K. Efficacy of Antibacterial Peptides Against Peptide-Resistant MRSA Is Restored by Permeabilization of Bacteria Membranes. Front. Microbiol. 2016, 7, 1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerhoff, H.V.; Juretić, D.; Hendler, R.W.; Zasloff, M. Magainins and the disruption of membrane-linked free-energy transduction. Proc. Natl. Acad. Sci. USA 1989, 86, 6597–6601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, D.; Nagarajan, T.; Roy, N.; Kulkarni, O.; Ravichandran, S.; Mishra, M.; Chakravortty, D.; Chandra, N. Computational antimicrobial peptide design and evaluation against multidrug-resistant clinical isolates of bacteria. J. Biol. Chem. 2018, 293, 3492–3509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, P.; El Amri, C. The dermaseptin superfamily: A gene-based combinatorial library of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1537–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castiglione-Morelli, M.A.; Cristinziano, P.; Pepe, A.; Temussi, P.A. Conformation-activity relationship of a novel peptide antibiotic: Structural characterization of dermaseptin DS 01 in media that mimic the membrane environment. Biopolymers 2005, 80, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Van Zoggel, H.; Carpentier, G.; Dos Santos, C.; Hamma-Kourbali, Y.; Courty, J.; Amiche, M.; Delbe, J. Antitumor and angiostatic activities of the antimicrobial peptide dermaseptin B2. PLoS ONE 2012, 7, e44351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irazazabal, L.N.; Porto, W.F.; Ribeiro, S.M.; Casale, S.; Humblot, V.; Ladram, A.; Franco, O.L. Selective amino acid substitution reduces cytotoxicity of the antimicrobial peptide mastoparan. Biochim. Biophys. Acta 2016, 1858, 2699–2708. [Google Scholar] [CrossRef]
- Navon-Venezia, S.; Feder, R.; Gaidukov, L.; Carmeli, Y.; Mor, A. Antibacterial properties of dermaseptin S4 derivatives with in vivo activity. Antimicrob. Agents Chemother. 2002, 46, 689–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krugliak, M.; Feder, R.; Zolotarev, V.Y.; Gaidukov, L.; Dagan, A.; Ginsburg, H.; Mor, A. Antimalarial activities of dermaseptin S4 derivatives. Antimicrob. Agents Chemother. 2000, 44, 2442–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, Y.; Wang, H.; Xi, X.; Ma, C.; Liu, Y.; Zhou, M.; Du, Q.; Burrows, J.F.; Wei, M.; Chen, T. Design of N-Terminal Derivatives from a Novel Dermaseptin Exhibiting Broad-Spectrum Antimicrobial Activity against Isolates from Cystic Fibrosis Patients. Biomolecules 2019, 9, 646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papo, N.; Oren, Z.; Pag, U.; Sahl, H.G.; Shai, Y. The consequence of sequence alteration of an amphipathic alpha-helical antimicrobial peptide and its diastereomers. J. Biol. Chem. 2002, 277, 33913–33921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.Y.; Oh, J.E.; Lee, K.H. Effect of D-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem. Pharmacol. 1999, 58, 1775–1780. [Google Scholar] [CrossRef]
- Braunstein, A.; Papo, N.; Shai, Y. In vitro activity and potency of an intravenously injected antimicrobial peptide and its DL amino acid analog in mice infected with bacteria. Antimicrob. Agents Chemother. 2004, 48, 3127–3129. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.L.; Nan, Y.H.; Hahm, K.S.; Shin, S.Y. Cell selectivity of an antimicrobial peptide melittin diastereomer with D-amino acid in the leucine zipper sequence. J. Biochem. Mol. Biol. 2007, 40, 1090–1094. [Google Scholar] [CrossRef] [Green Version]
- Westerhoff, H.V.; Hendler, R.W.; Zasloff, M.; Juretić, D. Interactions between a new class of eukaryotic antimicrobial agents and isolated rat liver mitochondria. Biochim. Biophys. Acta (BBA)-Bioenerg. 1989, 975, 361–369. [Google Scholar] [CrossRef]
- Bessalle, R.; Kapitkovsky, A.; Gorea, A.; Shalit, I.; Fridkin, M. All-D-magainin: Chirality, antimicrobial activity and proteolytic resistance. FEBS Lett. 1990, 274, 151–155. [Google Scholar]
- Juvvadi, P.; Vunnam, S.; Merrifield, R. Synthetic melittin, its enantio, retro, and retroenantio isomers, and selected chimeric analogs: Their antibacterial, hemolytic, and lipid bilayer action. J. Am. Chem. Soc. 1996, 118, 8989–8997. [Google Scholar] [CrossRef]
- Zhong, C.; Liu, T.; Gou, S.; He, Y.; Zhu, N.; Zhu, Y.; Wang, L.; Liu, H.; Zhang, Y.; Yao, J.; et al. Design and synthesis of new N-terminal fatty acid modified-antimicrobial peptide analogues with potent in vitro biological activity. Eur. J. Med. Chem. 2019, 182, 111636. [Google Scholar] [CrossRef]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Kim, J.; Huang, Z.; St Clair, J.R.; Brown, D.A.; London, E. Efficient replacement of plasma membrane outer leaflet phospholipids and sphingolipids in cells with exogenous lipids. Proc. Natl. Acad. Sci. USA 2016, 113, 14025–14030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.W. Peptide-lipid interactions and mechanisms of antimicrobial peptides. Novartis Found Symp. 1999, 225, 188–200, discussion 200–186. [Google Scholar] [PubMed]
- Almaaytah, A.; Farajallah, A.; Abualhaijaa, A.; Al-Balas, Q. A3, a Scorpion Venom Derived Peptide Analogue with Potent Antimicrobial and Potential Antibiofilm Activity against Clinical Isolates of Multi-Drug Resistant Gram Positive Bacteria. Molecules 2018, 23, 1603. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Li, Z.; Li, X.; Tian, Y.; Fan, Y.; Yu, C.; Zhou, B.; Liu, Y.; Xiang, R.; Yang, L. Synergistic effects of antimicrobial peptide DP7 combined with antibiotics against multidrug-resistant bacteria. Drug. Des. Devel. Ther. 2017, 11, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.J.; Hinner, M.J. Getting across the cell membrane: An overview for small molecules, peptides, and proteins. Methods Mol. Biol. 2015, 1266, 29–53. [Google Scholar] [CrossRef] [Green Version]
- Bryan, L.E.; Kwan, S. Roles of ribosomal binding, membrane potential, and electron transport in bacterial uptake of streptomycin and gentamicin. Antimicrob. Agents Chemother. 1983, 23, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Koga, K.; Kusawake, Y.; Ito, Y.; Sugioka, N.; Shibata, N.; Takada, K. Enhancing mechanism of Labrasol on intestinal membrane permeability of the hydrophilic drug gentamicin sulfate. Eur. J. Pharm. Biopharm. 2006, 64, 82–91. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, L.; Wang, J.; Wei, D.; Shi, B.; Shan, A. Synergistic interaction of PMAP-36 and PRW4 with aminoglycoside antibiotics and their antibacterial mechanism. World J. Microbiol. Biotechnol. 2014, 30, 3121–3128. [Google Scholar] [CrossRef]
- Liu, S. Characterizing the response of multidrug-resistant Klebsiella pneumoniae species to the application of a phage cocktail. Symposium 2014, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, G.A.; Munoz-Price, L.S. The new β-lactamases. N. Engl. J. Med. 2005, 352, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Van der Zwet, W.C.; Parlevliet, G.A.; Savelkoul, P.H.; Stoof, J.; Kaiser, A.M.; Koeleman, J.G.; Vandenbroucke-Grauls, C.M. Nosocomial outbreak of gentamicin-resistant Klebsiella pneumoniae in a neonatal intensive care unit controlled by a change in antibiotic policy. J. Hosp. Infect. 1999, 42, 295–302. [Google Scholar] [CrossRef]
- Van der Does, A.M.; Amatngalim, G.D.; Keijser, B.; Hiemstra, P.S.; Villenave, R. Contribution of Host Defence Proteins and Peptides to Host-Microbiota Interactions in Chronic Inflammatory Lung Diseases. Vaccines 2018, 6, 49. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wubbolts, R.W.; Haagsman, H.P.; Veldhuizen, E.J.A. Inhibition and Eradication of Pseudomonas aeruginosa Biofilms by Host Defence Peptides. Sci. Rep. 2018, 8, 10446. [Google Scholar] [CrossRef]
- Walter, R.; Neidle, A.; Marks, N. Significant differences in the degradation of pro-leu-gly-nH2 by human serum and that of other species (38484). Proc. Soc. Exp. Biol. Med. 1975, 148, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, M.; Qiu, S.; Wang, J.; Peng, J.; Zhao, P.; Zhu, R.; Wang, H.; Li, Y.; Wang, K.; et al. Antimicrobial activity and stability of the D-amino acid substituted derivatives of antimicrobial peptide polybia-MPI. AMB Express 2016, 6, 122. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.E.; Bevins, C.L. Paneth cells of the human small intestine express an antimicrobial peptide gene. J. Biol. Chem. 1992, 267, 23216–23225. [Google Scholar]
- Lehrer, R.I.; Lichtenstein, A.K.; Ganz, T. Defensins: Antimicrobial and cytotoxic peptides of mammalian cells. Annu. Rev. Immunol. 1993, 11, 105–128. [Google Scholar] [CrossRef]
- Li, W.-F.; Ma, G.-X.; Zhou, X.-X. Apidaecin-type peptides: Biodiversity, structure–function relationships and mode of action. Peptides 2006, 27, 2350–2359. [Google Scholar] [CrossRef]
- Jia, F.; Wang, J.; Peng, J.; Zhao, P.; Kong, Z.; Wang, K.; Yan, W.; Wang, R. D-amino acid substitution enhances the stability of antimicrobial peptide polybia-CP. Acta Biochim. Biophys. Sin. (Shanghai) 2017, 49, 916–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, T.; Liu, Y.; Tan, Z.; Ju, Y.; Yang, Y.; Dong, W. Antimicrobial activity, membrane interaction and stability of the D-amino acid substituted analogs of antimicrobial peptide W3R6. J. Photochem. Photobiol. B 2019, 200, 111645. [Google Scholar] [CrossRef] [PubMed]
- Oelkrug, C.; Hartke, M.; Schubert, A. Mode of action of anticancer peptides (ACPs) from amphibian origin. Anticancer Res. 2015, 35, 635–643. [Google Scholar] [PubMed]
- Lam, N.H.; Ma, Z.; Ha, B.Y. Electrostatic modification of the lipopolysaccharide layer: Competing effects of divalent cations and polycationic or polyanionic molecules. Soft Matter. 2014, 10, 7528–7544. [Google Scholar] [CrossRef]
- Murzyn, K.; Róg, T.; Pasenkiewicz-Gierula, M. Phosphatidylethanolamine-phosphatidylglycerol bilayer as a model of the inner bacterial membrane. Biophys. J. 2005, 88, 1091–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keil, B. Specificity of Proteolysis; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Carratalá, J.V.; Serna, N.; Villaverde, A.; Vázquez, E.; Ferrer-Miralles, N. Nanostructured antimicrobial peptides: The last push towards clinics. Biotechnol. Adv. 2020, 107603. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Combination effects of antimicrobial peptides. Antimicrob. Agents Chemother. 2016, 60, 1717–1724. [Google Scholar] [CrossRef] [Green Version]
- Yoshizawa, S.; Fourmy, D.; Puglisi, J.D. Structural origins of gentamicin antibiotic action. EMBO J. 1998, 17, 6437–6448. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, L.S., 3rd; Spencer, J.P. Aminoglycosides: A practical review. Am. Fam. Physician 1998, 58, 1811–1820. [Google Scholar]
- Kohanski, M.A.; Dwyer, D.J.; Wierzbowski, J.; Cottarel, G.; Collins, J.J. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 2008, 135, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Shan, A.; Ma, Z.; Xu, W.; Wang, J.; Chou, S.; Cheng, B. Bactericidal efficiency and modes of action of the novel antimicrobial peptide T9W against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2015, 59, 3008–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Zai, Y.; Xi, X.; Ma, C.; Wang, L.; Zhou, M.; Shaw, C.; Chen, T. A novel membrane-disruptive antimicrobial peptide from frog skin secretion against cystic fibrosis isolates and evaluation of anti-MRSA effect using Galleria mellonella model. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2019, 1863, 849–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tieleman, D.P.; Berendsen, H.J. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys. J. 1998, 74, 2786–2801. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Wu, D.; Xi, X.; Wu, Y.; Ma, C.; Zhou, M.; Wang, L.; Yang, M.; Chen, T.; Shaw, C. Identification and Characterisation of the Antimicrobial Peptide, Phylloseptin-PT, from the Skin Secretion of Phyllomedusa tarsius, and Comparison of Activity with Designed, Cationicity-Enhanced Analogues and Diastereomers. Molecules 2016, 21, 1667. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Chen, D.; Wang, L.; Lin, C.; Ma, C.; Xi, X.; Chen, T.; Shaw, C.; Zhou, M. Dermaseptin-PH: A Novel Peptide with Antimicrobial and Anticancer Activities from the Skin Secretion of the South American Orange-Legged Leaf Frog, Pithecopus (Phyllomedusa) hypochondrialis. Molecules 2017, 22, 1805. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L.; Wu, Y.; Wang, L.; Ma, C.; Xi, X.; Bininda-Emonds, O.R.P.; Shaw, C.; Chen, T.; Zhou, M. Evaluation of the bioactivity of a mastoparan peptide from wasp venom and of its analogues designed through targeted engineering. Int. J. Biol. Sci. 2018, 14, 599–607. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef]
- Hall, M.; Middleton, R.; Westmacott, D. The fractional inhibitory concentration (FIC) index as a measure of synergy. J. Antimicrob. Chemother. 1983, 11, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Desbois, A.P.; Coote, P.J. Wax moth larva (Galleria mellonella): An in vivo model for assessing the efficacy of antistaphylococcal agents. J. Antimicrob. Chemother. 2011, 66, 1785–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | MICs/MBCs (μM) | MICs (µg/mL)/(μM) | ||||
|---|---|---|---|---|---|---|
| DMPC-10B | DMPC-10A | Gentamicin | Vancomycin | Norfloxacin | ||
| Gram-positive bacteria | S. aureus (NCTC 10788) | 4/16 | 4/8 | <0.0625/<0.13 | 0.125/0.08 | 2/6.26 |
| MRSA (NCTC 12493) | 4/8 | 8/16 | 0.125/0.26 | 0.125/0.08 | 2/6.26 | |
| E. faecalis (NCTC 12697) | 64/64 | 64/64 | 4/8.38 | 1/0.69 | 4/12.53 | |
| Gram-negative bacteria | E. coli (NCTC 10418) | 2/4 | 8/8 | 1/2.09 | >32/>22.07 | 1/3.13 |
| K. pneumoniae (ATCC 43816) | 8/8 | 4/64 | 1/2.09 | >32/>22.07 | 2/6.26 | |
| K. pneumoniae (ATCC BAA 1705) | 32/32 | 32/32 | 2/4.19 | >32/>22.07 | >32/>100.21 | |
| K. pneumoniae (ATCC BAA 2342) | 16/16 | 16/16 | 4/8.38 | >32/>22.07 | >32/>100.21 | |
| P. aeruginosa (ATCC 27853) | 4/32 | 4/4 | 0.25/0.52 | >32/>22.07 | 2/6.26 | |
| Additive | Concentration | MICs of DMPC-10B (µM) | MICs of DMPC-10A (µM) | ||
|---|---|---|---|---|---|
| S. aureus | E. coli | S. aureus | E. coli | ||
| None | - | 4 | 2 | 4 | 8 |
| MgCl2 | 2 mM | 32 | 16 | 32 | 32 |
| 5 mM | 64 | 32 | 64 | 64 | |
| CaCl2 | 2 mM | 32 | 16 | 32 | 32 |
| 5 mM | 64 | 32 | 64 | 64 | |
| NaCl | 150 mM | 64 | 16 | 32 | 16 |
| 375 mM | 128 | 32 | 64 | 32 | |
| FBS | 10% | 8 | 4 | 16 | 16 |
| Combination | Bacteria Strains | ||
|---|---|---|---|
| K. pneumoniae (ATCC 43816) | K. pneumoniae (ATCC BAA 1705) | K. pneumoniae (ATCC BAA 2342) | |
| DMPC-10B | 1/8 | 4/32 | 2/16 |
| Gentamicin | 0.25/1 | 0.5/2 | 1/4 |
| FICI (DMPC-10B/Gentamicin) | 0.375 | 0.375 | 0.375 |
| DMPC-10B | 4/8 | 32/32 | 16/16 |
| Norfloxacin | 0.125/1 | 0.0625/>32 | 0.0625/>2 |
| FICI (DMPC-10B/Norfloxacin) | 0.625 | >1 | >1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zai, Y.; Ying, Y.; Ye, Z.; Zhou, M.; Ma, C.; Shi, Z.; Chen, X.; Xi, X.; Chen, T.; Wang, L. Broad-Spectrum Antimicrobial Activity and Improved Stability of a D-Amino Acid Enantiomer of DMPC-10A, the Designed Derivative of Dermaseptin Truncates. Antibiotics 2020, 9, 627. https://doi.org/10.3390/antibiotics9090627
Zai Y, Ying Y, Ye Z, Zhou M, Ma C, Shi Z, Chen X, Xi X, Chen T, Wang L. Broad-Spectrum Antimicrobial Activity and Improved Stability of a D-Amino Acid Enantiomer of DMPC-10A, the Designed Derivative of Dermaseptin Truncates. Antibiotics. 2020; 9(9):627. https://doi.org/10.3390/antibiotics9090627
Chicago/Turabian StyleZai, Yu, Yuan Ying, Zhuming Ye, Mei Zhou, Chengbang Ma, Zhanzhong Shi, Xiaoling Chen, Xinping Xi, Tianbao Chen, and Lei Wang. 2020. "Broad-Spectrum Antimicrobial Activity and Improved Stability of a D-Amino Acid Enantiomer of DMPC-10A, the Designed Derivative of Dermaseptin Truncates" Antibiotics 9, no. 9: 627. https://doi.org/10.3390/antibiotics9090627