Novel Antibiotics Targeting Bacterial Replicative DNA Polymerases


Abstract
:1. Introduction
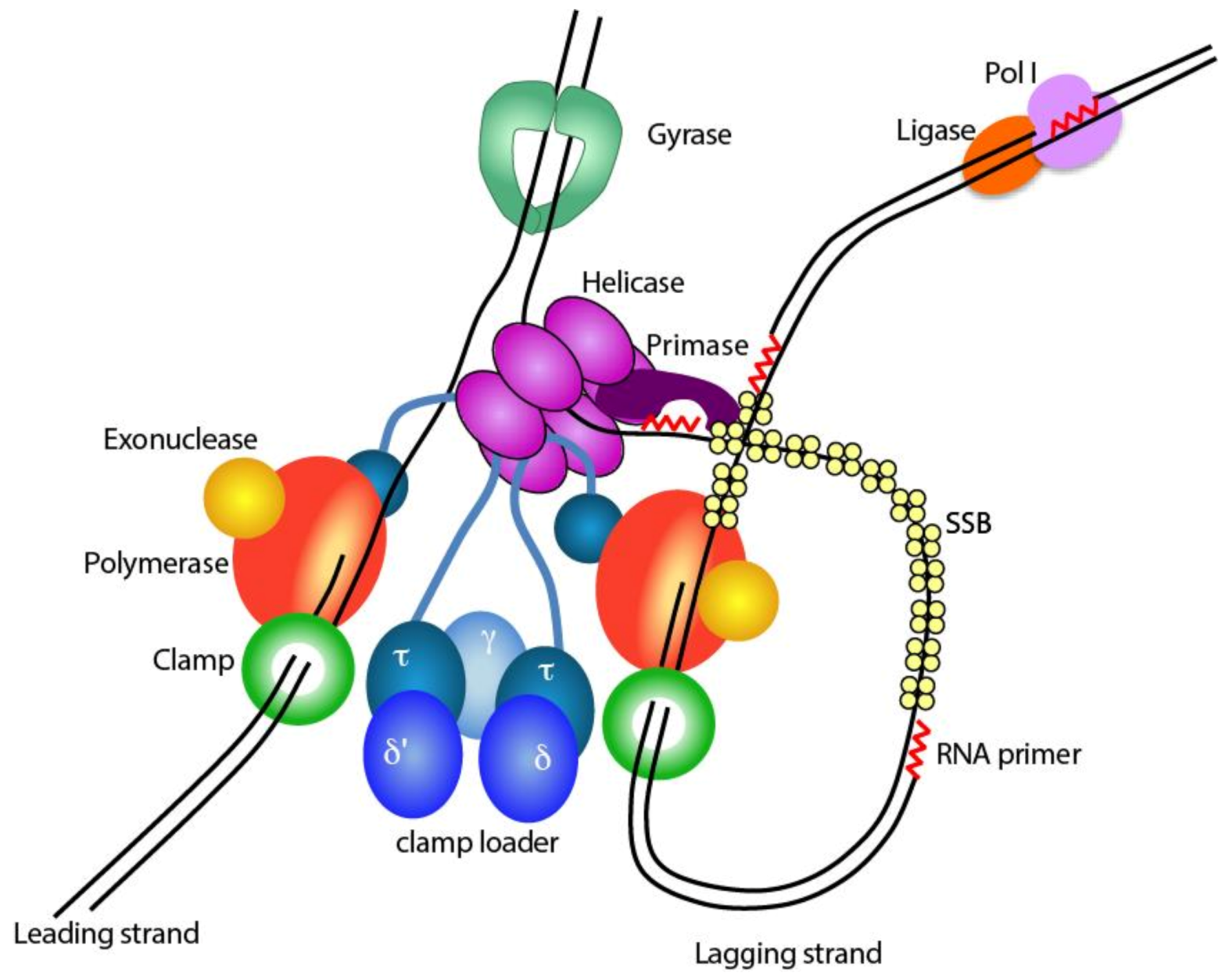
1.1. The Bacterial Replisome
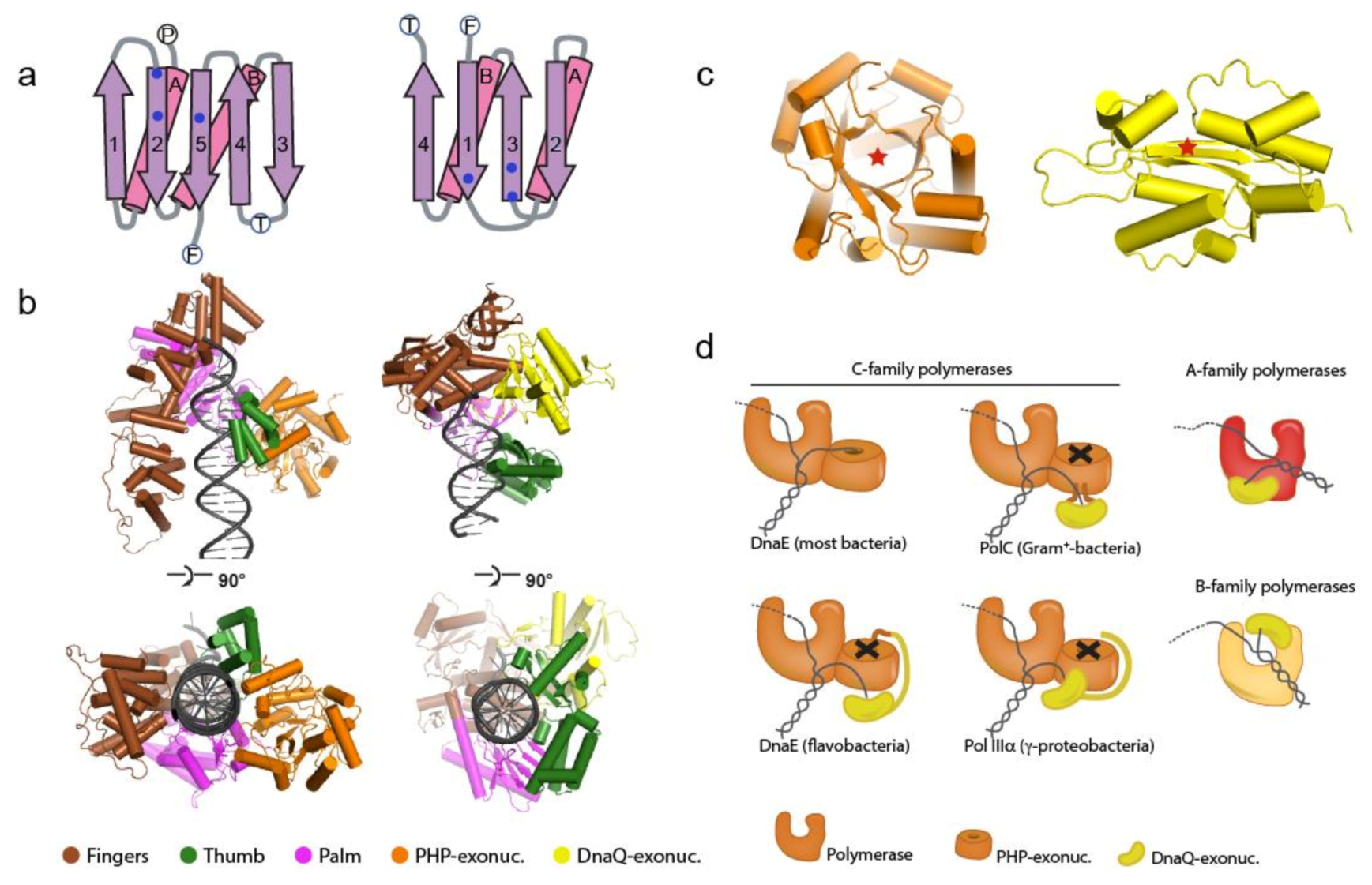
1.2. The Bacterial Replicative DNA Polymerases
2. Inhibiting Bacterial Replicative DNA Polymerases
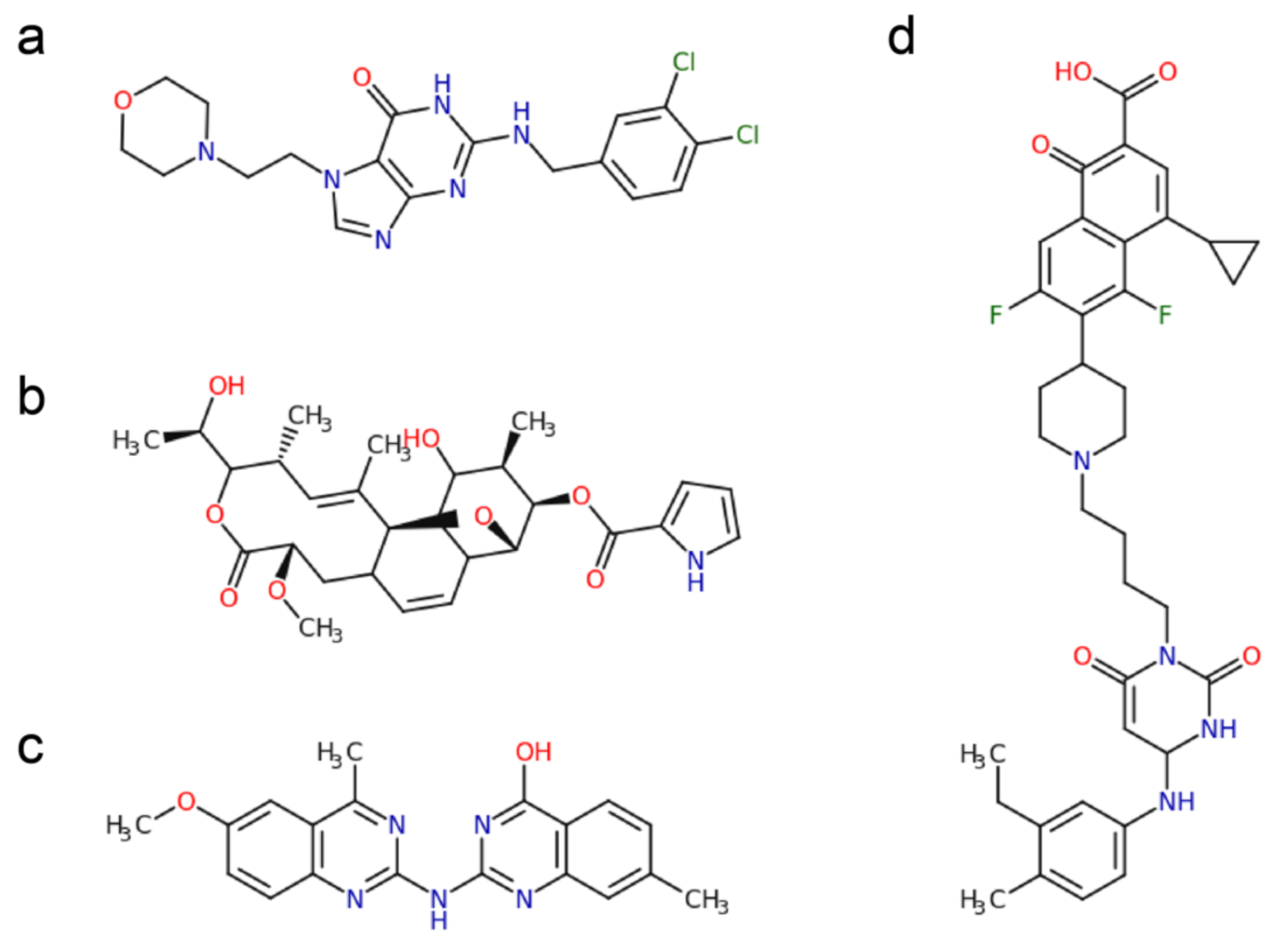
2.1. Polymerase Active Site Inhibitors
2.2. Allosteric Inhibition
2.3. Exonuclease Inhibitors
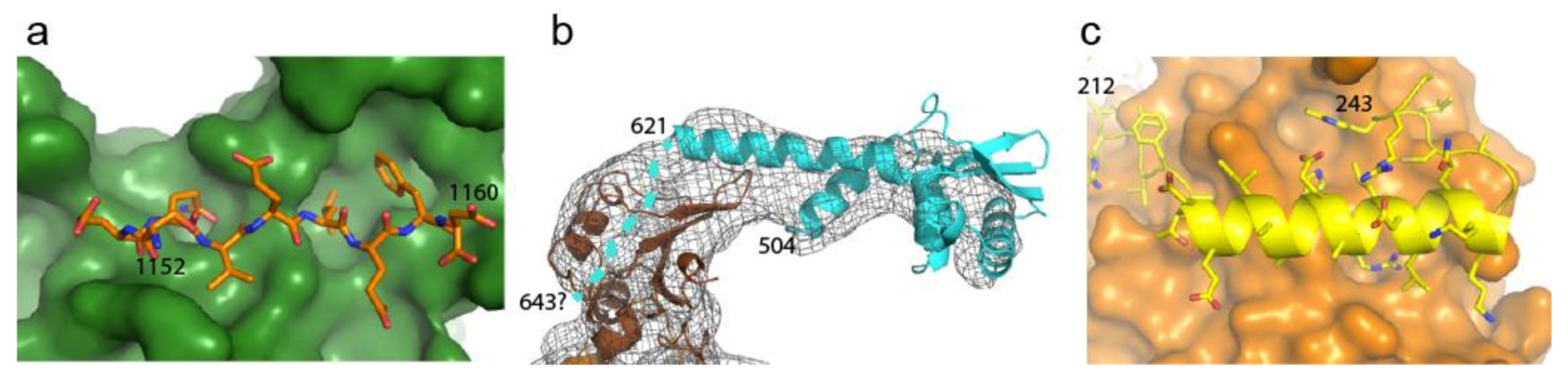
2.4. Disrupting Interaction with Replisome Proteins
2.5. Targeting DNA Polymerases to Combat Antibiotic Resistance
3. High-Throughput Screening Methods for Replication Inhibitors
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bbosa, G.S.; Mwebaza, N.; Odda, J.; Kyegombe, D.B.; Ntale, M. Antibiotics/antibacterial drug use, their marketing and promotion during the post-antibiotic golden age and their role in emergence of bacterial resistance. Health 2014, 6, 410–425. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, G.; Saigal, S.; Elongavan, A. Action and resistance mechanisms of antibiotics: A guide for clinicians. J. Anaesthesiol. Clin. Pharmacol. 2017, 33, 300–305. [Google Scholar] [CrossRef]
- Dallmann, H.G.; Fackelmayer, O.J.; Tomer, G.; Chen, J.; Wiktor-Becker, A.; Ferrara, T.; Pope, C.; Oliveira, M.T.; Burgers, P.M.J.; Kaguni, L.S.; et al. Parallel Multiplicative Target Screening against Divergent Bacterial Replicases: Identification of Specific Inhibitors with Broad Spectrum Potential. Biochemistry 2010, 49, 2551–2562. [Google Scholar] [CrossRef] [Green Version]
- Reiche, M.A.; Warner, D.F.; Mizrahi, V. Targeting DNA Replication and Repair for the Development of Novel Therapeutics against Tuberculosis. Front. Mol. Biosci. 2017, 4, 75. [Google Scholar] [CrossRef] [Green Version]
- Causer, R.J.; Dixon, N.E. Architecture and Conservation of the Bacterial DNA Replication Machinery, an Underexploited Drug Target. Curr. Drug Targets 2012, 13, 352–372. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, M.E.; Langston, L.; Stillman, B. Principles and Concepts of DNA Replication in Bacteria, Archaea, and Eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5, a010108. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.; O’Donnell, M. Cellular DNA Replicases: Components and Dynamics at the Replication Fork. Annu. Rev. Biochem. 2005, 74, 283–315. [Google Scholar] [CrossRef]
- Mok, M.; Marians, K.J. The Escherichia coli preprimosome and DNA B helicase can form replication forks that move at the same rate. J. Biol. Chem. 1987, 262, 16644–16654. [Google Scholar]
- Wu, A.C.; Zechner, E.L.; Marians, K.J. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. I. Multiple effectors act to modulate Okazaki fragment size. J. Biol. Chem. 1992, 267, 4030–4044. [Google Scholar] [PubMed]
- Lewis, J.S.; Spenkelink, L.M.; Jergic, S.; Wood, E.A.; Monachino, E.; Horan, N.P.; Duderstadt, K.E.; Cox, M.M.; Robinson, A.; Dixon, N.E.; et al. Single-molecule visualization of fast polymerase turnover in the bacterial replisome. eLife 2017, 6, e23932. [Google Scholar] [CrossRef] [PubMed]
- Tanner, A.N.; Hamdan, S.M.; Jergic, S.; Loscha, K.V.; Schaeffer, P.M.; Dixon, N.E.; Van Oijen, A.M. Single-molecule studies of fork dynamics in Escherichia coli DNA replication. Nat. Struct. Mol. Biol. 2008, 15, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Stukenberg, P.T.; Studwell-Vaughan, P.S.; O’Donnell, M. Mechanism of the sliding beta-clamp of DNA polymerase III holoenzyme. J. Biol. Chem. 1991, 266, 11328–11334. [Google Scholar]
- McHenry, C.S. Purification and characterization of DNA polymerase III’. Identification of tau as a subunit of the DNA polymerase III holoenzyme. J. Biol. Chem. 1982, 257, 2657–2663. [Google Scholar] [PubMed]
- Onrust, R.; Finkelstein, J.; Turner, J.; Naktinis, V.; O’Donnell, M. Assembly of a chromosomal replication machine: Two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. III. Interface between two polymerases and the clamp loader. J. Biol. Chem. 1995, 270, 13366–13377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, M.E. Accessory proteins bind a primed template and mediate rapid cycling of DNA polymerase III holoenzyme from Escherichia coli. J. Biol. Chem. 1987, 262, 16558–16565. [Google Scholar] [PubMed]
- Stukenberg, P.T.; Turner, J.; O’Donnell, M. An explanation for lagging strand replication: Polymerase hopping among DNA sliding clamps. Cell 1994, 78, 877–887. [Google Scholar] [CrossRef]
- McInerney, P.; Johnson, A.; Katz, F.; O’Donnell, M. Characterization of a Triple DNA Polymerase Replisome. Mol. Cell 2007, 27, 527–538. [Google Scholar] [CrossRef]
- Robinson, A.; Van Oijen, A.M. Bacterial replication, transcription and translation: Mechanistic insights from single-molecule biochemical studies. Nat. Rev. Genet. 2013, 11, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Licata, V.J. Pol I DNA polymerases stimulate DNA end-joining by Escherichia coli DNA ligase. Biochem. Biophys. Res. Commun. 2018, 497, 13–18. [Google Scholar] [CrossRef]
- Sissi, C.; Palumbo, M. In front of and behind the replication fork: Bacterial type IIA topoisomerases. Cell. Mol. Life Sci. 2010, 67, 2001–2024. [Google Scholar] [CrossRef]
- Sanyal, G.; Doig, P. Bacterial DNA replication enzymes as targets for antibacterial drug discovery. Expert Opin. Drug Discov. 2012, 7, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. MedChemComm 2019, 10, 1719–1739. [Google Scholar] [CrossRef]
- Kaguni, J.M. The Macromolecular Machines that Duplicate the Escherichia coli Chromosome as Targets for Drug Discovery. Antibiotics 2018, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Ito, J.; Braithwaite, D.K. Compilation and alignment of DNA polymerase sequences. Nucleic Acids Res. 1991, 19, 4045–4057. [Google Scholar] [CrossRef] [Green Version]
- Delarue, M.; Poch, O.; Tordo, N.; Moras, D.; Argos, P. An attempt to unify the structure of polymerases. Protein Eng. Des. Sel. 1990, 3, 461–467. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E.V. Archaeology of Eukaryotic DNA Replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a012963. [Google Scholar] [CrossRef]
- Bailey, S.; Wing, R.A.; Steitz, T.A. The Structure of T. aquaticus DNA Polymerase III is Distinct from Eukaryotic Replicative DNA Polymerases. Cell 2006, 126, 893–904. [Google Scholar] [CrossRef] [Green Version]
- Lamers, M.H.; Georgescu, R.E.; Lee, S.-G.; O’Donnell, M.; Kuriyan, J. Crystal Structure of the Catalytic α Subunit of E. coli Replicative DNA Polymerase III. Cell 2006, 126, 881–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravind, L.; Koonin, E.V. Phosphoesterase domains associated with DNA polymerases of diverse origins. Nucleic Acids Res. 1998, 26, 3746–3752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rock, J.M.; Lang, U.F.; Chase, M.R.; Ford, C.B.; Gerrick, E.R.; Gawande, R.; Coscolla, M.; Gagneux, S.; Fortune, S.M.; Lamers, M.H. DNA replication fidelity in Mycobacterium tuberculosis is mediated by an ancestral prokaryotic proofreader. Nat. Genet. 2015, 47, 677–681. [Google Scholar] [CrossRef]
- Timinskas, K.; Balvočiūtė, M.; Timinskas, A.; Venclovas, Č. Comprehensive analysis of DNA polymerase III α subunits and their homologs in bacterial genomes. Nucleic Acids Res. 2013, 42, 1393–1413. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef]
- Zarrouk, K.; Piret, J.; Boivin, G. Herpesvirus DNA polymerases: Structures, functions and inhibitors. Virus Res. 2017, 234, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Fischl, M.A.; Richman, D.D.; Grieco, M.H.; Gottlieb, M.S.; Volberding, P.A.; Laskin, O.L.; Leedom, J.M.; Groopman, J.E.; Mildvan, D.; Schooley, R.T.; et al. The Efficacy of Azidothymidine (AZT) in the Treatment of Patients with AIDS and AIDS-Related Complex. N. Engl. J. Med. 1987, 317, 185–191. [Google Scholar] [CrossRef]
- Tempestilli, M.; Caputi, P.; Avataneo, V.; Notari, S.; Forini, O.; Scorzolini, L.; Marchioni, L.; Bartoli, T.A.; Castilletti, C.; Lalle, E.; et al. Pharmacokinetics of remdesivir and GS-441524 in two critically ill patients who recovered from COVID-19. J. Antimicrob. Chemother. 2020, 75, 2977–2980. [Google Scholar] [CrossRef] [PubMed]
- Vashishtha, A.K.; Kuchta, R.D. Effects of Acyclovir, Foscarnet, and Ribonucleotides on Herpes Simplex Virus-1 DNA Polymerase: Mechanistic Insights and a Novel Mechanism for Preventing Stable Incorporation of Ribonucleotides into DNA. Biochemistry 2016, 55, 1168–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gool, I.C.; Rayner, E.; Osse, E.M.; Nout, R.A.; Creutzberg, C.L.; Tomlinson, I.; Church, D.N.; Smit, V.T.; De Wind, N.; Bosse, T.; et al. Adjuvant Treatment forPOLEProofreading Domain–Mutant Cancers: Sensitivity to Radiotherapy, Chemotherapy, and Nucleoside Analogues. Clin. Cancer Res. 2018, 24, 3197–3203. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; De Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056. [Google Scholar] [CrossRef]
- Painter, R.E.; Adam, G.C.; Arocho, M.; DiNunzio, E.; Donald, R.G.; Dorso, K.; Genilloud, O.; Gill, C.; Goetz, M.; Hairston, N.N.; et al. Elucidation of DnaE as the Antibacterial Target of the Natural Product, Nargenicin. Chem. Biol. 2015, 22, 1362–1373. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.-C.; Silverman, M.H.; Yu, X.Y.; Wright, G.; Brown, N. Discovery and development of DNA polymerase IIIC inhibitors to treat Gram-positive infections. Bioorganic Med. Chem. 2019, 27, 3209–3217. [Google Scholar] [CrossRef]
- Mondal, S.I.; Ferdous, S.; Akter, A.; Mahmud, Z.; Karim, N.; Islam, M.S.; Jewel, N.A.; Afrin, T. Identification of potential drug targets by subtractive genome analysis of Escherichia coli O157:H7: An in silico approach. Adv. Appl. Bioinform. Chem. 2015, 8, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Z.; Zhou, Y.; Li, J.; Zhang, X.; Shi, X.; Xue, X.; Li, Z.; Ma, B.; Wang, Y.; Li, M.; et al. Selective in vivo and in vitro activities of 3,3′-4-nitrobenzylidene-bis-4-hydroxycoumarin against methicillin-resistant Staphylococcus aureus by inhibition of DNA polymerase III. Sci. Rep. 2015, 5, 13637. [Google Scholar] [CrossRef]
- Jadaun, A.; Sudhakar, D.R.; Subbarao, N.; Dixit, A. In silico screening for novel inhibitors of DNA polymerase III alpha subunit of Mycobacterium tuberculosis (MtbDnaE2, H37Rv). PLoS ONE 2015, 10, e0119760. [Google Scholar]
- Barnes, M.H.; Butler, M.M.; Wright, G.E.; Brown, N.C. Antimicrobials targeted to the replication-specific DNA polymerases of gram-positive bacteria: Target potential of dnaE. Infect. Disord. Drug Targets 2012, 12, 327–331. [Google Scholar] [CrossRef]
- Xu, W.-C.; Wright, G.E.; Brown, N.C.; Long, Z.-Y.; Zhi, C.-X.; Dvoskin, S.; Gambino, J.J.; Barnes, M.H.; Butler, M.M. 7-Alkyl-N(2)-substituted-3-deazaguanines. Synthesis, DNA polymerase III inhibition and antibacterial activity. Bioorganic Med. Chem. Lett. 2011, 21, 4197–4202. [Google Scholar] [CrossRef] [Green Version]
- Guiles, J.W.; Sun, X.; Critchley, I.A.; Ochsner, U.; Tregay, M.; Stone, K.; Bertino, J.; Green, L.; Sabin, R.; Dean, F.B.; et al. Quinazolin-2-ylamino-quinazolin-4-ols as novel non-nucleoside inhibitors of bacterial DNA polymerase III. Bioorganic Med. Chem. Lett. 2009, 19, 800–802. [Google Scholar] [CrossRef]
- Butler, M.M.; LaMarr, W.A.; Foster, K.A.; Barnes, M.H.; Skow, D.J.; Lyden, P.T.; Kustigian, L.M.; Zhi, C.; Brown, N.C.; Wright, G.E.; et al. Antibacterial Activity and Mechanism of Action of a Novel Anilinouracil-Fluoroquinolone Hybrid Compound. Antimicrob. Agents Chemother. 2007, 51, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarantino, P.M.; Zhi, C.; Gambino, J.J.; Wright, G.E.; Brown, N.C. 6-Anilinouracil-Based Inhibitors of Bacillus subtilis DNA Polymerase III: Antipolymerase and Antimicrobial Structure−Activity Relationships Based on Substitution at Uracil N3. J. Med. Chem. 1999, 42, 2035–2040. [Google Scholar] [CrossRef]
- Corona, A.; Masaoka, T.; Tocco, G.; Tramontano, E.; Le Grice, S.F.J. Active site and allosteric inhibitors of the ribonuclease H activity of HIV reverse transcriptase. Futur. Med. Chem. 2013, 5, 2127–2139. [Google Scholar] [CrossRef]
- Kohlstaedt, A.L.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Bird, L.; Chamberlain, P.P.; Stewart-Jones, G.B.; Stuart, D.I.; Stammers, D.K. Structure of HIV-2 reverse transcriptase at 2.35-A resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc. Natl. Acad. Sci. USA 2002, 99, 14410–14415. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Leiro, R.; Conrad, J.; Scheres, S.H.W.; Lamers, M.H. Cryo-EM structures of the E. coli replicative DNA polymerase reveal its dynamic interactions with the DNA sliding clamp, exonuclease and τ. eLife 2015, 4, e11134. [Google Scholar] [CrossRef]
- Fernandez-Leiro, R.; Conrad, J.; Yang, J.-C.; Freund, S.M.V.; Scheres, S.H.W.; Lamers, M.H. Self-correcting mismatches during high-fidelity DNA replication. Nat. Struct. Mol. Biol. 2017, 24, 140–143. [Google Scholar] [CrossRef]
- Bębenek, A.; Ziuzia-Graczyk, I. Fidelity of DNA replication—A matter of proofreading. Curr. Genet. 2018, 64, 985–996. [Google Scholar] [CrossRef] [Green Version]
- Brutlag, D.; Kornberg, A. Enzymatic synthesis of deoxyribonucleic acid. 36. A proofreading function for the 3′ leads to 5′ exonuclease activity in deoxyribonucleic acid polymerases. J. Biol. Chem. 1972, 247, 241–248. [Google Scholar]
- Reha-Krantz, L.J.; Marquez-Curtis, L.; Elisseeva, E.; Baker, R.P.; Bloom, L.B.; Dunford, H.B.; Goodman, M.F. The Proofreading Pathway of Bacteriophage T4 DNA Polymerase. J. Biol. Chem. 1998, 273, 22969–22976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, R.P.; Reha-Krantz, L.J. Identification of a transient excision intermediate at the crossroads between DNA polymerase extension and proofreading pathways. Proc. Natl. Acad. Sci. USA 1998, 95, 3507–3512. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, E.F.; Reha-Krantz, L.J. DNA polymerase proofreading: Active site switching catalyzed by the bacteriophage T4 DNA polymerase. Nucleic Acids Res. 2007, 35, 5452–5463. [Google Scholar] [CrossRef]
- Lancy, E.D.; Lifsics, M.R.; Kehres, D.G.; Maurer, R. Isolation and characterization of mutants with deletions in dnaQ, the gene for the editing subunit of DNA polymerase III in Salmonella typhimurium. J. Bacteriol. 1989, 171, 5572–5580. [Google Scholar] [CrossRef] [Green Version]
- Scheuermann, R.; Tam, S.; Burgers, P.M.; Lu, C.; Echols, H. Identification of the epsilon-subunit of Escherichia coli DNA polymerase III holoenzyme as the dnaQ gene product: A fidelity subunit for DNA replication. Proc. Natl. Acad. Sci. USA 1983, 80, 7085–7089. [Google Scholar] [CrossRef] [Green Version]
- Reardon, E.J.; Spector, T. Herpes simplex virus type 1 DNA polymerase. Mechanism of inhibition by acyclovir triphosphate. J. Biol. Chem. 1989, 264, 7405–7411. [Google Scholar]
- Hamdan, S.; Bulloch, E.M.; Thompson, P.R.; Beck, J.L.; Yang, J.Y.; Crowther, J.A.; Lilley, P.E.; Carr, P.D.; Ollis, D.L.; Brown, S.E.; et al. Hydrolysis of the 5′-p-nitrophenyl ester of TMP by the proofreading exonuclease (epsilon) subunit of Escherichia coli DNA polymerase III. Biochemistry 2002, 41, 5266–5275. [Google Scholar] [PubMed]
- Standish, A.J.; Salim, A.A.; Capon, R.J.; Morona, R. Dual inhibition of DNA polymerase PolC and protein tyrosine phosphatase CpsB uncovers a novel antibiotic target. Biochem. Biophys. Res. Commun. 2013, 430, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Banos-Mateos, S.; Van Roon, A.-M.M.; Lang, U.F.; Maslen, S.L.; Skehel, J.M.; Lamers, M.H. High-fidelity DNA replication in Mycobacterium tuberculosis relies on a trinuclear zinc center. Nat. Commun. 2017, 8, 855. [Google Scholar] [CrossRef] [Green Version]
- Nasir, N.; Kisker, C. Mechanistic insights into the enzymatic activity and inhibition of the replicative polymerase exonuclease domain from Mycobacterium tuberculosis. DNA Repair 2019, 74, 17–25. [Google Scholar] [CrossRef]
- Cihlar, T.; Fordyce, M. Current status and prospects of HIV treatment. Curr. Opin. Virol. 2016, 18, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, N.Y.; Georgescu, R.E.; Finkelstein, J.; O’Donnell, M.E. Single-molecule analysis reveals that the lagging strand increases replisome processivity but slows replication fork progression. Proc. Natl. Acad. Sci. USA 2009, 106, 13236–13241. [Google Scholar] [CrossRef] [Green Version]
- Wijffels, G.; Dalrymple, B.P.; Prosselkov, P.; Kongsuwan, K.; Epa, V.C.; Lilley, P.E.; Jergic, S.; Buchardt, J.; Brown, S.E.; Alewood, P.F.; et al. Inhibition of protein interactions with the beta 2 sliding clamp of Escherichia coli DNA polymerase III by peptides from beta 2-binding proteins. Biochemistry 2004, 43, 5661–5671. [Google Scholar]
- Georgescu, R.E.; Yurieva, O.; Kim, S.-S.; Kuriyan, J.; Kong, X.-P.; O’Donnell, M. Structure of a small-molecule inhibitor of a DNA polymerase sliding clamp. Proc. Natl. Acad. Sci. USA 2008, 105, 11116–11121. [Google Scholar] [CrossRef] [Green Version]
- Kling, A.; Lukat, P.; Almeida, D.V.; Bauer, A.; Fontaine, E.; Sordello, S.; Zaburannyi, N.; Herrmann, J.; Wenzel, S.C.; König, C.; et al. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 2015, 348, 1106–1112. [Google Scholar] [CrossRef]
- Duderstadt, K.E.; Geertsema, H.J.; Stratmann, S.A.; Punter, C.M.; Kulczyk, A.W.; Richardson, C.C.; Van Oijen, A.M. Simultaneous Real-Time Imaging of Leading and Lagging Strand Synthesis Reveals the Coordination Dynamics of Single Replisomes. Mol. Cell 2016, 64, 1035–1047. [Google Scholar] [CrossRef] [Green Version]
- Jergic, S.; Ozawa, K.; Williams, N.K.; Su, X.C.; Scott, D.D.; Hamdan, S.M.; Crowther, J.A.; Otting, G.; Dixon, N.E. The unstructured C-terminus of the tau subunit of Escherichia coli DNA polymerase III holoenzyme is the site of interaction with the alpha subunit. Nucleic Acids Res. 2007, 35, 2813–2824. [Google Scholar] [PubMed] [Green Version]
- Su, X.C.; Jergic, S.; Keniry, M.A.; Dixon, N.E.; Otting, G. Solution structure of Domains IVa and V of the tau subunit of Escherichia coli DNA polymerase III and interaction with the alpha subunit. Nucleic Acids Res. 2007, 35, 2825–2832. [Google Scholar]
- Perrino, F.W.; Harvey, S.; McNeill, S.M. Two functional domains of the epsilon subunit of DNA polymerase III. Biochemistry 1999, 38, 16001–16009. [Google Scholar]
- Ozawa, K.; Horan, N.P.; Robinson, A.; Yagi, H.; Hill, F.R.; Jergic, S.; Xu, Z.-Q.; Loscha, K.V.; Li, N.; Tehei, M.; et al. Proofreading exonuclease on a tether: The complex between the E. coli DNA polymerase III subunits α, ε, θ and β reveals a highly flexible arrangement of the proofreading domain. Nucleic Acids Res. 2013, 41, 5354–5367. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Wang, Y.; Whittell, L.R.; Jergic, S.; Liu, M.; Harry, E.; Dixon, N.E.; Kelso, M.J.; Beck, J.L.; Oakley, A.J. DNA Replication Is the Target for the Antibacterial Effects of Nonsteroidal Anti-Inflammatory Drugs. Chem. Biol. 2014, 21, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef]
- Warner, D.F.; Rock, J.M.; Fortune, S.M.; Mizrahi, V. DNA Replication Fidelity in the Mycobacterium tuberculosis Complex. Adv. Exp. Med. Biol. 2017, 247–262. [Google Scholar] [CrossRef]
- Sutton, M.D.; Walker, G.C. Managing DNA polymerases: Coordinating DNA replication, DNA repair, and DNA recombination. Proc. Natl. Acad. Sci. USA 2001, 98, 8342–8349. [Google Scholar] [CrossRef] [Green Version]
- Boshoff, H.I.; Reed, M.B.; Barry, C.E.; Mizrahi, V. DnaE2 Polymerase Contributes to In Vivo Survival and the Emergence of Drug Resistance in Mycobacterium tuberculosis. Cell 2003, 113, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Zhang, C.; Zhao, M. Singly labeled smart probes for real-time monitoring of the kinetics of dNTP misincorporation and single nucleotide extension in DNA intra-molecular polymerization. Biosens. Bioelectron. 2009, 25, 301–305. [Google Scholar] [CrossRef]
- Rêgo, A.T.; Holding, A.N.; Kent, H.; Lamers, M.H. Architecture of the Pol III–clamp–exonuclease complex reveals key roles of the exonuclease subunit in processive DNA synthesis and repair. EMBO J. 2013, 32, 1334–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, A.B.; Rivin, O.; Gao, N.; Hajec, L. A homogeneous, high-throughput fluorescence resonance energy transfer-based DNA polymerase assay. Anal. Biochem. 2005, 347, 254–261. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Nucleotide | Polymerase | Species | Reference |
|---|---|---|---|---|
| ACX-362E 1 | Yes | PolC | C. difficile | [41] |
| ACX-375C | Yes | PolC | S. aureus, others | Acurxpharma 3 |
| Nargenicin | No | DnaE | S. aureus | [40] |
| DB04118, DB04698 2 | No | Pol IIIα | E. coli | [42] |
| NBH | No | PolC | S. aureus | [43] |
| Multiple 2 | Yes, No | DnaE2 4 | M. tuberculosis | [44] |
| 324C | Yes | DnaE | B. subtilis | [45] |
| 7-Alkyl-N(2)-substituted-3-deazaguanines | Yes | PolC, Pol IIIα | B. subtilis | [46] |
| quinazolin-2-ylamino-quinazolin-4-ols | No | PolC | S. aureus | [47] |
| 251D | No | PolC | B. subtilis | [48] |
| 6-Anilinouracil-based | Yes | PolC | B. subtilis | [49] |
| Compound | Target Site | Polymerase | Species | Reference |
|---|---|---|---|---|
| dADP, dCDP, dGDP | PHP | DnaE1 | M. tuberculosis | [66] |
| fascioquinol | exo-PHP | PolC | S. pneumoniae | [64] |
| Compound | Target Site | Polymerase | Species | Reference |
|---|---|---|---|---|
| Griselimycin | clamp | DnaE1 | M. tuberculosis | [71] |
| Vedaprofen, bromfenac and carprofen | clamp | Pol IIIα | E. coli, S. aureus, B. subtilis | [77] |
| RU7 1 | clamp | Pol IIIα | E. coli | [70] |
| Peptides | clamp | Pol IIIα | E. coli | [69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, J.A.; Lamers, M.H. Novel Antibiotics Targeting Bacterial Replicative DNA Polymerases. Antibiotics 2020, 9, 776. https://doi.org/10.3390/antibiotics9110776
Santos JA, Lamers MH. Novel Antibiotics Targeting Bacterial Replicative DNA Polymerases. Antibiotics. 2020; 9(11):776. https://doi.org/10.3390/antibiotics9110776
Chicago/Turabian StyleSantos, Joana A., and Meindert H. Lamers. 2020. "Novel Antibiotics Targeting Bacterial Replicative DNA Polymerases" Antibiotics 9, no. 11: 776. https://doi.org/10.3390/antibiotics9110776