Blocking the Trigger: Inhibition of the Initiation of Bacterial Chromosome Replication as an Antimicrobial Strategy

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Proteins Involved in Initiation of Bacterial Chromosome Replication
2.1. Overview
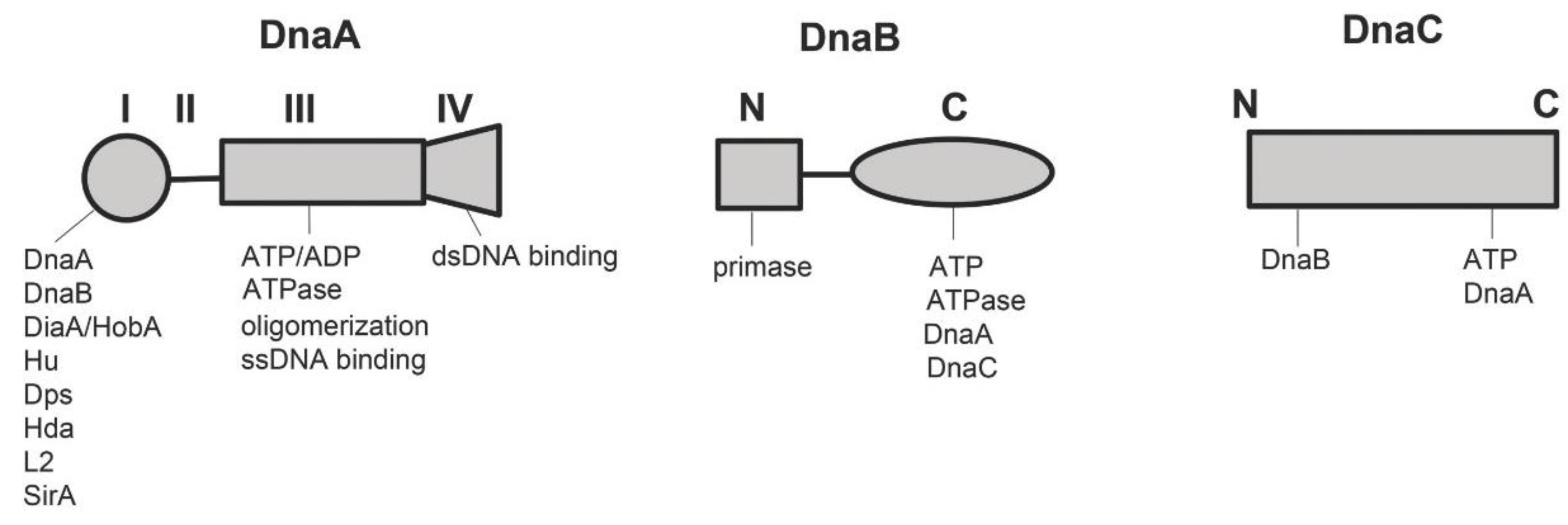
2.1.1. DnaA
2.1.2. DnaB and DnaC
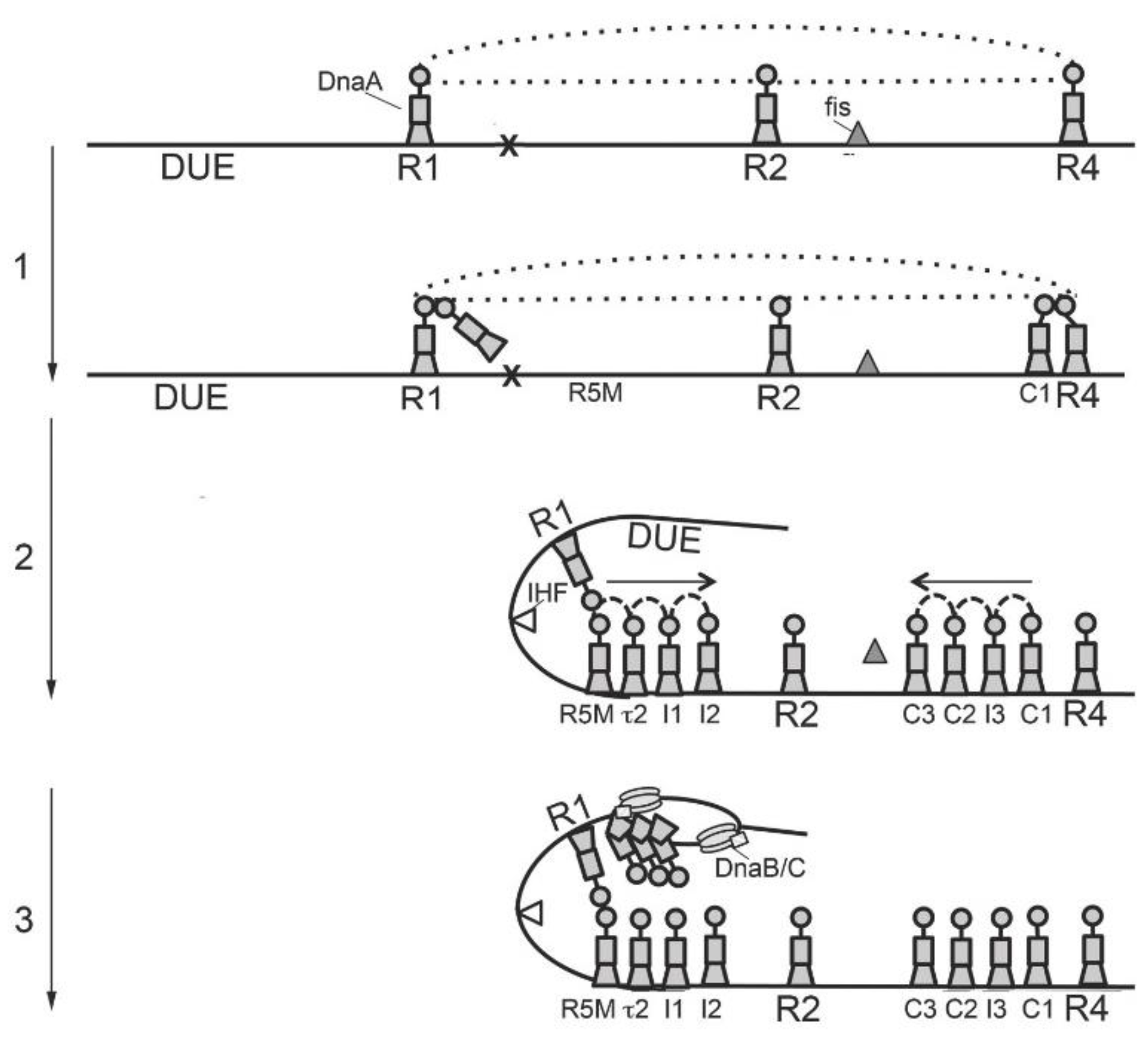
3. Orisome Assembly
4. Inhibitors of Replication Initiation: Where We are and Where We might Go
4.1. Overview
4.2. In Vitro Screens Targeting DnaA and DnaB
4.3. Cell Based Screen for Inhibitors of Bacterial Replication Initiation
4.4. New Strategies for Inhibiting Initiation Based on Orisome Assembly and Regulation Pathways
Author Contributions
Funding
Conflicts of Interest
References
- Davies, J. Where have All the Antibiotics Gone. Can. J. Infect. Dis Med. Microbiol. 2006, 17, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. PT 2015, 40, 277–283. [Google Scholar]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Agersø, Y.; Bjerre, K.; Brockmann, E.; Johansen, E.; Nielsen, B.; Siezen, R.; Stuer-Lauridsen, B.; Wels, M.; Zeidan, A.A. Putative antibiotic resistance genes present in extant Bacillus licheniformis and Bacillus paralicheniformis strains are probably intrinsic and part of the ancient resistome. PLoS ONE 2019, 14, e0210363. [Google Scholar] [CrossRef]
- Jiang, X.L.; Ellabaan, M.M.H.; Charusanti, P.; Munck, C.; Blin, K.; Tong, Y.J.; Weber, T.; Sommer, M.O.A.; Lee, S.Y. Dissemination of antibiotic resistance genes from antibiotic producers to pathogens. Nat. Commun. 2017, 8, 15784. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, R.; Davies, J. Aminoglycoside antibiotic-inactivating enzymes in actinomycetes similar to those present in clinical isolates of antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. USA 1973, 70, 2276–2280. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.; Kaur, P. Antibiotic Resistance Mechanisms in Bacteria: Relationships Between Resistance Determinants of Antibiotic Producers, Environmental Bacteria, and Clinical Pathogens. Front. Microbiol. 2018, 9, 2928. [Google Scholar] [CrossRef] [PubMed]
- Talbot, G.H.; Jezek, A.; Murray, B.E.; Jones, R.N.; Ebright, R.H.; Nau, G.J.; Rodvold, K.A.; Newland, J.G.; Boucher, H.W. The Infectious Diseases Society of America’s 10ב20 Initiative (Ten New Systemic Antibacterial Agents FDA-approved by 2020): Is 20ב20 a Possibility. Clin. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Mori, H.; Baba, T.; Yokoyama, K.; Takeuchi, R.; Nomura, W.; Makishi, K.; Otsuka, Y.; Dose, H.; Wanner, B.L. Identification of essential genes and synthetic lethal gene combinations in Escherichia coli K-12. Methods Mol. Biol. 2015, 1279, 45–65. [Google Scholar] [CrossRef]
- Glass, J.I.; Merryman, C.; Wise, K.S.; Hutchison, C.A.; Smith, H.O. Minimal Cells-Real and Imagined. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Sekimizu, K.; Bramhill, D.; Kornberg, A. Sequential early stages in the in vitro initiation of replication at the origin of the Escherichia coli chromosome. J. Biol. Chem. 1988, 263, 7124–7130. [Google Scholar] [PubMed]
- Løbner-Olesen, A.; Skarstad, K.; Hansen, F.G.; von Meyenburg, K.; Boye, E. The DnaA protein determines the initiation mass of Escherichia coli K-12. Cell 1989, 57, 881–889. [Google Scholar] [CrossRef]
- Messer, W.; Blaesing, F.; Jakimowicz, D.; Krause, M.; Majka, J.; Nardmann, J.; Schaper, S.; Seitz, H.; Speck, C.; Weigel, C. Bacterial replication initiator DnaA. Rules for DnaA binding and roles of DnaA in origin unwinding and helicase loading. Biochimie 2001, 83, 5–12. [Google Scholar] [CrossRef]
- Kaguni, J.M. DnaA: Controlling the initiation of bacterial DNA replication and more. Annu. Rev. Microbiol. 2006, 60, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Orlova, N.; Gerding, M.; Ivashkiv, O.; Olinares, P.D.B.; Chait, B.T.; Waldor, M.K.; Jeruzalmi, D. The replication initiator of the cholera pathogen’s second chromosome shows structural similarity to plasmid initiators. Nucleic Acids Res. 2017, 45, 3724–3737. [Google Scholar] [CrossRef] [PubMed]
- Bramhill, D.; Kornberg, A. Duplex opening by dnaA protein at novel sequences in initiation of replication at the origin of the E. coli chromosome. Cell 1988, 52, 743–755. [Google Scholar] [CrossRef]
- Ishida, T.; Akimitsu, N.; Kashioka, T.; Hatano, M.; Kubota, T.; Ogata, Y.; Sekimizu, K.; Katayama, T. DiaA, a novel DnaA-binding protein, ensures the timely initiation of Escherichia coli chromosome replication. J. Biol. Chem. 2004, 279, 45546–45555. [Google Scholar] [CrossRef]
- Terradot, L.; Zawilak-Pawlik, A. Structural insight into Helicobacter pylori DNA replication initiation. Gut Microbes 2010, 1, 330–334. [Google Scholar] [CrossRef]
- Chodavarapu, S.; Felczak, M.M.; Yaniv, J.R.; Kaguni, J.M. Escherichia coli DnaA interacts with HU in initiation at the E. coli replication origin. Mol. Microbiol. 2008, 67, 781–792. [Google Scholar] [CrossRef]
- Chodavarapu, S.; Gomez, R.; Vicente, M.; Kaguni, J.M. Escherichia coli Dps interacts with DnaA protein to impede initiation: A model of adaptive mutation. Mol. Microbiol. 2008, 67, 1331–1346. [Google Scholar] [CrossRef] [PubMed]
- Su’etsugu, M.; Harada, Y.; Keyamura, K.; Matsunaga, C.; Kasho, K.; Abe, Y.; Ueda, T.; Katayama, T. The DnaA N-terminal domain interacts with Hda to facilitate replicase clamp-mediated inactivation of DnaA. Environ. Microbiol. 2013, 15, 3183–3195. [Google Scholar] [CrossRef] [PubMed]
- Chodavarapu, S.; Felczak, M.M.; Kaguni, J.M. Two forms of ribosomal protein L2 of Escherichia coli that inhibit DnaA in DNA replication. Nucleic Acids Res. 2011, 39, 4180–4191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahn-Lee, L.; Merrikh, H.; Grossman, A.D.; Losick, R. The sporulation protein SirA inhibits the binding of DnaA to the origin of replication by contacting a patch of clustered amino acids. J. Bacteriol. 2011, 193, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Chodavarapu, S.; Kaguni, J.M. Replication Initiation in Bacteria. Enzymes 2016, 39, 1–30. [Google Scholar] [Green Version]
- Smits, W.K.; Goranov, A.I.; Grossman, A.D. Ordered association of helicase loader proteins with the Bacillus subtilis origin of replication in vivo. Mol. Microbiol. 2010, 75, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Soultanas, P. Loading mechanisms of ring helicases at replication origins. Mol. Microbiol. 2012, 84, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Simmons, L.A.; Felczak, M.; Kaguni, J.M. DnaA Protein of Escherichia coli: Oligomerization at the E. coli chromosomal origin is required for initiation and involves specific N-terminal amino acids. Mol. Microbiol. 2003, 49, 849–858. [Google Scholar] [CrossRef]
- Zawilak-Pawlik, A.; Nowaczyk, M.; Zakrzewska-Czerwińska, J. The Role of the N-Terminal Domains of Bacterial Initiator DnaA in the Assembly and Regulation of the Bacterial Replication Initiation Complex. Genes 2017, 8, 136. [Google Scholar] [CrossRef]
- Miller, D.T.; Grimwade, J.E.; Betteridge, T.; Rozgaja, T.; Torgue, J.J.; Leonard, A.C. Bacterial origin recognition complexes direct assembly of higher-order DnaA oligomeric structures. Proc. Natl. Acad. Sci. USA 2009, 106, 18479–18484. [Google Scholar] [CrossRef] [Green Version]
- Rozgaja, T.A.; Grimwade, J.E.; Iqbal, M.; Czerwonka, C.; Vora, M.; Leonard, A.C. Two oppositely oriented arrays of low-affinity recognition sites in oriC guide progressive binding of DnaA during Escherichia coli pre-RC assembly. Mol. Microbiol. 2011, 82, 475–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molt, K.L.; Sutera, V.A.; Moore, K.K.; Lovett, S.T. A role for nonessential domain II of initiator protein, DnaA, in replication control. Genetics 2009, 183, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Katayama, T. DnaA, ORC, and Cdc6: Similarity beyond the domains of life and diversity. Biochem. Cell Biol. 2010, 88, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Erzberger, J.P.; Pirruccello, M.M.; Berger, J.M. The structure of bacterial DnaA: Implications for general mechanisms underlying DNA replication initiation. EMBO J. 2002, 21, 4763–4773. [Google Scholar] [CrossRef] [PubMed]
- Duderstadt, K.E.; Berger, J.M. AAA+ ATPases in the initiation of DNA replication. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T.; Kasho, K.; Kawakami, H. The DnaA Cycle in Escherichia coli: Activation, Function and Inactivation of the Initiator Protein. Front. Microbiol. 2017, 8, 2496. [Google Scholar] [CrossRef] [PubMed]
- Yung, B.Y.; Crooke, E.; Kornberg, A. Fate of the DnaA initiator protein in replication at the origin of the Escherichia coli chromosome in vitro. J. Biol. Chem. 1990, 265, 1282–1285. [Google Scholar] [PubMed]
- Grimwade, J.E.; Torgue, J.J.; McGarry, K.C.; Rozgaja, T.; Enloe, S.T.; Leonard, A.C. Mutational analysis reveals Escherichia coli oriC interacts with both DnaA-ATP and DnaA-ADP during pre-RC assembly. Mol. Microbiol. 2007, 66, 428–439. [Google Scholar] [CrossRef]
- Grimwade, J.E.; Rozgaja, T.A.; Gupta, R.; Dyson, K.; Rao, P.; Leonard, A.C. Origin recognition is the predominant role for DnaA-ATP in initiation of chromosome replication. Nucleic Acids Res. 2018, 46, 6140–6151. [Google Scholar] [CrossRef]
- Mukhopadhyay, G.; Carr, K.M.; Kaguni, J.M.; Chattoraj, D.K. Open-complex formation by the host initiator, DnaA, at the origin of P1 plasmid replication. EMBO J. 1993, 12, 4547–4554. [Google Scholar] [CrossRef]
- McGarry, K.C.; Ryan, V.T.; Grimwade, J.E.; Leonard, A.C. Two discriminatory binding sites in the Escherichia coli replication origin are required for DNA strand opening by initiator DnaA-ATP. Proc. Natl. Acad. Sci. USA 2004, 101, 2811–2816. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Keyamura, K.; Katayama, T. Formation of an ATP-DnaA-specific initiation complex requires DnaA Arginine 285, a conserved motif in the AAA+ protein family. J. Biol. Chem. 2005, 280, 27420–27430. [Google Scholar] [CrossRef] [PubMed]
- Carr, K.M.; Kaguni, J.M. The A184V missense mutation of the dnaA5 and dnaA46 alleles confers a defect in ATP binding and thermolability in initiation of Escherichia coli DNA replication. Mol. Microbiol. 1996, 20, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Duderstadt, K.E.; Mott, M.L.; Crisona, N.J.; Chuang, K.; Yang, H.; Berger, J.M. Origin remodeling and opening in bacteria rely on distinct assembly states of the DnaA initiator. J. Biol. Chem. 2010, 285, 28229–28239. [Google Scholar] [CrossRef] [PubMed]
- Felczak, M.M.; Kaguni, J.M. The box VII motif of Escherichia coli DnaA protein is required for DnaA oligomerization at the E. coli replication origin. J. Biol. Chem. 2004, 279, 51156–51162. [Google Scholar] [CrossRef] [PubMed]
- Erzberger, J.P.; Mott, M.L.; Berger, J.M. Structural basis for ATP-dependent DnaA assembly and replication-origin remodeling. Nat. Struct. Mol. Biol. 2006, 13, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.; Noguchi, Y.; Hayashi, Y.; Miyazaki, E.; Katayama, T. Differentiation of the DnaA-oriC subcomplex for DNA unwinding in a replication initiation complex. J. Biol. Chem. 2012, 287, 37458–37471. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.T.; Harran, O.; Murray, H. The bacterial DnaA-trio replication origin element specifies single-stranded DNA initiator binding. Nature 2016, 534, 412–416. [Google Scholar] [CrossRef]
- Fuller, R.S.; Funnell, B.E.; Kornberg, A. The dnaA protein complex with the E. coli chromosomal replication origin (oriC) and other DNA sites. Cell 1984, 38, 889–900. [Google Scholar] [CrossRef]
- Fujikawa, N.; Kurumizaka, H.; Nureki, O.; Terada, T.; Shirouzu, M.; Katayama, T.; Yokoyama, S. Structural basis of replication origin recognition by the DnaA protein. Nucleic Acids Res. 2003, 31, 2077–2086. [Google Scholar] [CrossRef] [Green Version]
- Grimwade, J.E.; Ryan, V.T.; Leonard, A.C. IHF redistributes bound initiator protein, DnaA, on supercoiled oriC of Escherichia coli. Mol. Microbiol. 2000, 35, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Schaper, S.; Messer, W. Interaction of the initiator protein DnaA of Escherichia coli with its DNA target. J. Biol. Chem. 1995, 270, 17622–17626. [Google Scholar] [CrossRef] [PubMed]
- Charbon, G.; Lobner-Olesen, A. A role for the weak DnaA binding sites in bacterial replication origins. Mol. Microbiol. 2011, 82, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.A.; Ouimet, M.C.; Wargachuk, R.; Marczynski, G.T. The Caulobacter crescentus chromosome replication origin evolved two classes of weak DnaA binding sites. Mol. Microbiol. 2011, 82, 312–326. [Google Scholar] [CrossRef]
- Donczew, R.; Mielke, T.; Jaworski, P.; Zakrzewska-Czerwińska, J.; Zawilak-Pawlik, A. Assembly of Helicobacter pylori initiation complex is determined by sequence-specific and topology-sensitive DnaA-oriC interactions. J. Mol. Biol. 2014, 426, 2769–2782. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Nakai, S.; Moriya, S.; Yoshikawa, H.; Ogasawara, N. The Bacillus subtilis dnaC gene encodes a protein homologous to the DnaB helicase of Escherichia coli. Microbiology 1995, 141, 641–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galletto, R.; Jezewska, M.J.; Bujalowski, W. Interactions of the Escherichia coli DnaB helicase hexamer with the replication factor the DnaC protein. Effect of nucleotide cofactors and the ssDNA on protein-protein interactions and the topology of the complex. J. Mol. Biol. 2003, 329, 441–465. [Google Scholar] [CrossRef]
- Li, Y.; Araki, H. Loading and activation of DNA replicative helicases: The key step of initiation of DNA replication. Genes Cells 2013, 18, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Wahle, E.; Lasken, R.S.; Kornberg, A. The dnaB-dnaC replication protein complex of Escherichia coli. II. Role of the complex in mobilizing dnaB functions. J. Biol. Chem. 1989, 264, 2469–2475. [Google Scholar]
- Arias-Palomo, E.; O’Shea, V.L.; Hood, I.V.; Berger, J.M. The bacterial DnaC helicase loader is a DnaB ring breaker. Cell 2013, 153, 438–448. [Google Scholar] [CrossRef]
- Arias-Palomo, E.; Puri, N.; O’Shea Murray, V.L.; Yan, Q.; Berger, J.M. Physical Basis for the Loading of a Bacterial Replicative Helicase onto DNA. Mol. Cell 2019, 74, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.D.; Carr, K.M.; Vicente, M.; Kaguni, J.M. Escherichia coli DnaA protein. The N-terminal domain and loading of DnaB helicase at the E. coli chromosomal origin. J. Biol. Chem. 1998, 273, 34255–34262. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.; Weigel, C.; Messer, W. The interaction domains of the DnaA and DnaB replication proteins of Escherichia coli. Mol. Microbiol. 2000, 37, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, Y.; Nishimura, M.; Hayashi, C.; Akama, Y.; Ozaki, S.; Katayama, T. The DnaA AAA+ Domain His136 Residue Directs DnaB Replicative Helicase to the Unwound Region of the Replication Origin, oriC. Front. Microbiol. 2018, 9, 2017. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.J.; Fang, L.; McInerney, P.; Georgescu, R.E.; O’Donnell, M. The DnaC helicase loader is a dual ATP/ADP switch protein. EMBO J. 2002, 21, 3148–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiasa, H.; Marians, K.J. Initiation of bidirectional replication at the chromosomal origin is directed by the interaction between helicase and primase. J. Biol. Chem. 1999, 274, 27244–27248. [Google Scholar] [CrossRef]
- Corn, J.E.; Berger, J.M. Regulation of bacterial priming and daughter strand synthesis through helicase-primase interactions. Nucleic Acids Res. 2006, 34, 4082–4088. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Dallmann, H.G.; McHenry, C.S.; Marians, K.J. Coupling of a replicative polymerase and helicase: A tau-DnaB interaction mediates rapid replication fork movement. Cell 1996, 84, 643–650. [Google Scholar] [CrossRef]
- Baker, T.A.; Funnell, B.E.; Kornberg, A. Helicase action of dnaB protein during replication from the Escherichia coli chromosomal origin in vitro. J. Biol. Chem. 1987, 262, 6877–6885. [Google Scholar]
- Mott, M.L.; Erzberger, J.P.; Coons, M.M.; Berger, J.M. Structural synergy and molecular crosstalk between bacterial helicase loaders and replication initiators. Cell 2008, 135, 623–634. [Google Scholar] [CrossRef]
- Smits, W.K.; Merrikh, H.; Bonilla, C.Y.; Grossman, A.D. Primosomal proteins DnaD and DnaB are recruited to chromosomal regions bound by DnaA in Bacillus subtilis. J. Bacteriol. 2011, 193, 640–648. [Google Scholar] [CrossRef]
- Caspi, R.; Pacek, M.; Consiglieri, G.; Helinski, D.R.; Toukdarian, A.; Konieczny, I. A broad host range replicon with different requirements for replication initiation in three bacterial species. EMBO J. 2001, 20, 3262–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, R.K.; Mehra, P.; Mukhopadhyay, G.D.; Har, S.K. Helicobacter pylori DnaB helicase can bypass Escherichia coli DnaC function in vivo. Biochem. J. 2005, 389, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Brézellec, P.; Petit, M.A.; Pasek, S.; Vallet-Gely, I.; Possoz, C.; Ferat, J.L. Domestication of Lambda Phage Genes into a Putative Third Type of Replicative Helicase Matchmaker. Genome Biol. Evol. 2017, 9, 1561–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawilak-Pawlik, A.; Kois, A.; Majka, J.; Jakimowicz, D.; Smulczyk-Krawczyszyn, A.; Messer, W.; Zakrzewska-Czerwińska, J. Architecture of bacterial replication initiation complexes: Orisomes from four unrelated bacteria. Biochem. J. 2005, 389, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.C.; Grimwade, J.E. Regulation of DnaA assembly and activity: Taking directions from the genome. Annu. Rev. Microbiol. 2011, 65, 19–35. [Google Scholar] [CrossRef]
- Leonard, A.C.; Grimwade, J.E. The orisome: Structure and function. Front. Microbiol. 2015, 6, 545. [Google Scholar] [CrossRef]
- Samitt, C.E.; Hansen, F.G.; Miller, J.F.; Schaechter, M. In vivo studies of DnaA binding to the origin of replication of Escherichia coli. EMBO J. 1989, 8, 989–993. [Google Scholar] [CrossRef]
- Cassler, M.R.; Grimwade, J.E.; Leonard, A.C. Cell cycle-specific changes in nucleoprotein complexes at a chromosomal replication origin. EMBO J. 1995, 14, 5833–5841. [Google Scholar] [CrossRef]
- Nievera, C.; Torgue, J.J.; Grimwade, J.E.; Leonard, A.C. SeqA blocking of DnaA-oriC interactions ensures staged assembly of the E. coli pre-RC. Mol. Cell 2006, 24, 581–592. [Google Scholar] [CrossRef]
- Kaur, G.; Vora, M.P.; Czerwonka, C.A.; Rozgaja, T.A.; Grimwade, J.E.; Leonard, A.C. Building the bacterial orisome: High-affinity DnaA recognition plays a role in setting the conformation of oriC DNA. Mol. Microbiol. 2014, 91, 1148–1163. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, Y.; Sakiyama, Y.; Kawakami, H.; Katayama, T. The Arg Fingers of Key DnaA Protomers Are Oriented Inward within the Replication Origin oriC and Stimulate DnaA Subcomplexes in the Initiation Complex. J. Biol. Chem. 2015, 290, 20295–20312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurokawa, K.; Nishida, S.; Emoto, A.; Sekimizu, K.; Katayama, T. Replication cycle-coordinated change of the adenine nucleotide-bound forms of DnaA protein in Escherichia coli. EMBO J. 1999, 18, 6642–6652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duderstadt, K.E.; Chuang, K.; Berger, J.M. DNA stretching by bacterial initiators promotes replication origin opening. Nature 2011, 478, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Duderstadt, K.E.; Berger, J.M. A structural framework for replication origin opening by AAA+ initiation factors. Curr. Opin. Struct. Biol. 2013, 23, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.; Kawakami, H.; Nakamura, K.; Fujikawa, N.; Kagawa, W.; Park, S.Y.; Yokoyama, S.; Kurumizaka, H.; Katayama, T. A common mechanism for the ATP-DnaA-dependent formation of open complexes at the replication origin. J. Biol. Chem. 2008, 283, 8351–8362. [Google Scholar] [CrossRef] [PubMed]
- Speck, C.; Messer, W. Mechanism of origin unwinding: Sequential binding of DnaA to double-and single-stranded DNA. EMBO J. 2001, 20, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Yung, B.Y.; Kornberg, A. The dnaA initiator protein binds separate domains in the replication origin of Escherichia coli. J. Biol. Chem. 1989, 264, 6146–6150. [Google Scholar] [PubMed]
- Ozaki, S.; Katayama, T. Highly organized DnaA-oriC complexes recruit the single-stranded DNA for replication initiation. Nucleic Acids Res. 2012, 40, 1648–1665. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Campbell, J.L.; Boye, E.; Kleckner, N. SeqA: A negative modulator of replication initiation in E. COLI. Cell 1994, 77, 413–426. [Google Scholar] [CrossRef]
- Katayama, T.; Sekimizu, K. Inactivation of Escherichia coli DnaA protein by DNA polymerase III and negative regulations for initiation of chromosomal replication. Biochimie 1999, 81, 835–840. [Google Scholar] [CrossRef]
- Skarstad, K.; Katayama, T. Regulating DNA replication in bacteria. Cold Spring Harb. Perspect. Biol. 2013, 5, a012922. [Google Scholar] [CrossRef] [PubMed]
- Wolański, M.; Donczew, R.; Zawilak-Pawlik, A.; Zakrzewska-Czerwińska, J. oriC-encoded instructions for the initiation of bacterial chromosome replication. Front. Microbiol. 2014, 5, 735. [Google Scholar] [CrossRef] [PubMed]
- Speck, C.; Weigel, C.; Messer, W. From footprint to toeprint: A close-up of the DnaA box, the binding site for the bacterial initiator protein DnaA. Nucleic Acids Res. 1997, 25, 3242–3247. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.C.; Méchali, M. DNA replication origins. Cold Spring Harb. Perspect. Biol. 2013, 5, a010116. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Gao, F. DoriC 10.0: An updated database of replication origins in prokaryotic genomes including chromosomes and plasmids. Nucleic Acids Res. 2019, 47, D74–D77. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Rückert, B.; Lurz, R.; Messer, W. Complexes at the replication origin of Bacillus subtilis with homologous and heterologous DnaA protein. J. Mol. Biol. 1997, 274, 365–380. [Google Scholar] [CrossRef]
- Jakimowicz, D.; Majkadagger, J.; Konopa, G.; Wegrzyn, G.; Messer, W.; Schrempf, H.; Zakrzewska-Czerwińska, J. Architecture of the Streptomyces lividans DnaA protein-replication origin complexes. J. Mol. Biol. 2000, 298, 351–364. [Google Scholar] [CrossRef]
- Robinson, A.; Causer, R.J.; Dixon, N.E. Architecture and conservation of the bacterial DNA replication machinery, an underexploited drug target. Curr. Drug Targets 2012, 13, 352–372. [Google Scholar] [CrossRef]
- Fossum, S.; De Pascale, G.; Weigel, C.; Messer, W.; Donadio, S.; Skarstad, K. A robust screen for novel antibiotics: Specific knockout of the initiator of bacterial DNA replication. FEMS Microbiol. Lett. 2008, 281, 210–214. [Google Scholar] [CrossRef]
- Klitgaard, R.N.; Lobner-Olesen, A. A Novel Fluorescence Based Screen for Inhibitors of the Initiation of DNA Replication in Bacteria. Curr. Drug Discov. Technol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yamaichi, Y.; Duigou, S.; Shakhnovich, E.A.; Waldor, M.K. Targeting the replication initiator of the second Vibrio chromosome: Towards generation of vibrionaceae-specific antimicrobial agents. PLoS Pathog. 2009, 5, e1000663. [Google Scholar] [CrossRef] [PubMed]
- Schallopp, N.; Milbredt, S.; Sperlea, T.; Kemter, F.S.; Bruhn, M.; Schindler, D.; Waldminghaus, T. Establishing a System for Testing Replication Inhibition of the Vibrio cholerae Secondary Chromosome in Escherichia coli. Antibiotics 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Charbon, G.; Klitgaard, R.N.; Liboriussen, C.D.; Thulstrup, P.W.; Maffioli, S.I.; Donadio, S.; Løbner-Olesen, A. Iron chelation increases the tolerance of Escherichia coli to hyper-replication stress. Sci. Rep. 2018, 8, 10550. [Google Scholar] [CrossRef] [PubMed]
- Sekimizu, K.; Bramhill, D.; Kornberg, A. ATP activates dnaA protein in initiating replication of plasmids bearing the origin of the E. coli chromosome. Cell 1987, 50, 259–265. [Google Scholar] [CrossRef]
- Mizushima, T.; Sasaki, S.; Ohishi, H.; Kobayashi, M.; Katayama, T.; Miki, T.; Maeda, M.; Sekimizu, K. Molecular design of inhibitors of in vitro oriC DNA replication based on the potential to block the ATP binding of DnaA protein. J. Biol. Chem. 1996, 271, 25178–25183. [Google Scholar] [CrossRef] [PubMed]
- Kaguni, J.M. The Macromolecular Machines that Duplicate the Escherichia coli Chromosome as Targets for Drug Discovery. Antibiotics 2018, 7, 23. [Google Scholar] [CrossRef]
- Li, B.; Pai, R.; Aiello, D.; Di, M.; Barnes, M.H.; Peet, N.P.; Bowlin, T.L.; Moir, D.T. Optimization of a novel potent and selective bacterial DNA helicase inhibitor scaffold from a high throughput screening hit. Bioorg. Med. Chem Lett. 2013, 23, 3481–3486. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Huang, C.Y. Inhibition of Klebsiella pneumoniae DnaB helicase by the flavonol galangin. Protein J. 2011, 30, 59–65. [Google Scholar] [CrossRef]
- Griep, M.A.; Blood, S.; Larson, M.A.; Koepsell, S.A.; Hinrichs, S.H. Myricetin inhibits Escherichia coli DnaB helicase but not primase. Bioorg. Med. Chem. 2007, 15, 7203–7208. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.H.; Huang, C.Y. Characterization of flavonol inhibition of DnaB helicase: Real-time monitoring, structural modeling, and proposed mechanism. J. Biomed. Biotechnol. 2012, 2012, 735368. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Pai, R.; Di, M.; Aiello, D.; Barnes, M.H.; Butler, M.M.; Tashjian, T.F.; Peet, N.P.; Bowlin, T.L.; Moir, D.T. Coumarin-based inhibitors of Bacillus anthracis and Staphylococcus aureus replicative DNA helicase: Chemical optimization, biological evaluation, and antibacterial activities. J. Med. Chem. 2012, 55, 10896–10908. [Google Scholar] [CrossRef] [PubMed]
- McKay, G.A.; Reddy, R.; Arhin, F.; Belley, A.; Lehoux, D.; Moeck, G.; Sarmiento, I.; Parr, T.R.; Gros, P.; Pelletier, J. Triaminotriazine DNA helicase inhibitors with antibacterial activity. Bioorg. Med. Chem. Lett. 2006, 16, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Venkova-Canova, T.; Srivastava, P.; Chattoraj, D.K. Transcriptional inactivation of a regulatory site for replication of Vibrio cholerae chromosome II. Proc. Natl. Acad. Sci. USA 2006, 103, 12051–12056. [Google Scholar] [CrossRef] [PubMed]
- Weigel, C.; Messer, W.; Preiss, S.; Welzeck, M.; MorigenBoye, E. The sequence requirements for a functional Escherichia coli replication origin are different for the chromosome and a minichromosome. Mol. Microbiol. 2001, 40, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Kogoma, T. Stable DNA replication: Interplay between DNA replication, homologous recombination, and transcription. Microbiol. Mol. Biol. Rev. 1997, 61, 212–238. [Google Scholar] [PubMed]
- Simmons, L.A.; Breier, A.M.; Cozzarelli, N.R.; Kaguni, J.M. Hyperinitiation of DNA replication in Escherichia coli leads to replication fork collapse and inviability. Mol. Microbiol. 2004, 51, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Simmons, L.A.; Kaguni, J.M. The DnaAcos allele of Escherichia coli: Hyperactive initiation is caused by substitution of A184V and Y271H, resulting in defective ATP binding and aberrant DNA replication control. Mol. Microbiol. 2003, 47, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Charbon, G.; Bjørn, L.; Mendoza-Chamizo, B.; Frimodt-Møller, J.; Løbner-Olesen, A. Oxidative DNA damage is instrumental in hyperreplication stress-induced inviability of Escherichia coli. Nucleic Acids Res. 2014, 42, 13228–13241. [Google Scholar] [CrossRef] [PubMed]
- Charbon, G.; Riber, L.; Løbner-Olesen, A. Countermeasures to survive excessive chromosome replication in Escherichia coli. Curr. Genet. 2018, 64, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, L.; Weigel, C.; von Kries, J.; Møller, M.; Skarstad, K. A novel DNA gyrase inhibitor rescues Escherichia coli dnaAcos mutant cells from lethal hyperinitiation. J. Antimicrob. Chemother. 2010, 65, 924–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Freiesleben, U.; Rasmussen, K.V. DNA replication in Escherichia coli gyrB(Ts) mutants analysed by flow cytometry. Res. Microbiol. 1991, 142, 223–227. [Google Scholar] [CrossRef]
- Imlay, J.A. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat. Rev. Microbiol. 2013, 11, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.C.; Grimwade, J.E. Building a bacterial orisome: Emergence of new regulatory features for replication origin unwinding. Mol. Microbiol. 2005, 55, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Ryan, V.T.; Grimwade, J.E.; Camara, J.E.; Crooke, E.; Leonard, A.C. Escherichia coli prereplication complex assembly is regulated by dynamic interplay among Fis, IHF and DnaA. Mol. Microbiol. 2004, 51, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.; Rozgaja, T.A.; Alqahtani, A.; Grimwade, J.E.; Leonard, A.C. Low Affinity DnaA-ATP Recognition Sites in E. coli oriC Make Non-equivalent and Growth Rate-Dependent Contributions to the Regulated Timing of Chromosome Replication. Front. Microbiol. 2018, 9, 1673. [Google Scholar] [CrossRef] [PubMed]
- Merrikh, H.; Grossman, A.D. Control of the replication initiator DnaA by an anti-cooperativity factor. Mol. Microbiol. 2011, 82, 434–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholefield, G.; Whiting, R.; Errington, J.; Murray, H. Spo0J regulates the oligomeric state of Soj to trigger its switch from an activator to an inhibitor of DNA replication initiation. Mol. Microbiol. 2011, 79, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Scholefield, G.; Murray, H. YabA and DnaD inhibit helix assembly of the DNA replication initiation protein DnaA. Mol. Microbiol. 2013, 90, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Scholefield, G.; Errington, J.; Murray, H. Soj/ParA stalls DNA replication by inhibiting helix formation of the initiator protein DnaA. EMBO J. 2012, 31, 1542–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonilla, C.Y.; Grossman, A.D. The primosomal protein DnaD inhibits cooperative DNA binding by the replication initiator DnaA in Bacillus subtilis. J. Bacteriol. 2012, 194, 5110–5117. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, G.; Sarva, K.B.; Blaszczyk, E.; Rajagopalan, M.; Madiraju, M.V. Mycobacterium tuberculosis oriC sequestration by MtrA response regulator. Mol. Microbiol. 2015, 98, 586–604. [Google Scholar] [CrossRef] [PubMed]
- Donczew, R.; Makowski, Ł.; Jaworski, P.; Bezulska, M.; Nowaczyk, M.; Zakrzewska-Czerwińska, J.; Zawilak-Pawlik, A. The atypical response regulator HP1021 controls formation of the Helicobacter pylori replication initiation complex. Mol. Microbiol. 2015, 95, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Mognol, G.P.; González-Avalos, E.; Ghosh, S.; Spreafico, R.; Gudlur, A.; Rao, A.; Damoiseaux, R.; Hogan, P.G. Targeting the NFAT:AP-1 transcriptional complex on DNA with a small-molecule inhibitor. Proc. Natl. Acad. Sci. USA 2019, 116, 9959–9968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gormally, M.V.; Dexheimer, T.S.; Marsico, G.; Sanders, D.A.; Lowe, C.; Matak-Vinković, D.; Michael, S.; Jadhav, A.; Rai, G.; Maloney, D.J. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat. Commun. 2014, 5, 5165. [Google Scholar] [CrossRef] [PubMed]
- Grimley, E.; Liao, C.; Ranghini, E.J.; Nikolovska-Coleska, Z.; Dressler, G.R. Inhibition of Pax2 Transcription Activation with a Small Molecule that Targets the DNA Binding Domain. ACS Chem. Biol. 2017, 12, 724–734. [Google Scholar] [CrossRef]
- Kjelstrup, S.; Hansen, P.M.; Thomsen, L.E.; Hansen, P.R.; Løbner-Olesen, A. Cyclic peptide inhibitors of the β-sliding clamp in Staphylococcus aureus. PLoS ONE 2013, 8, e72273. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Whittell, L.R.; Wang, Y.; Jergic, S.; Ma, C.; Lewis, P.J.; Dixon, N.E.; Beck, J.L.; Kelso, M.J.; Oakley, A.J. Bacterial Sliding Clamp Inhibitors that Mimic the Sequential Binding Mechanism of Endogenous Linear Motifs. J. Med. Chem. 2015, 58, 4693–4702. [Google Scholar] [CrossRef] [Green Version]
- Georgescu, R.E.; Yurieva, O.; Kim, S.S.; Kuriyan, J.; Kong, X.P.; O’Donnell, M. Structure of a small-molecule inhibitor of a DNA polymerase sliding clamp. Proc. Natl. Acad. Sci. USA 2008, 105, 11116–11121. [Google Scholar] [CrossRef] [Green Version]
- Kling, A.; Lukat, P.; Almeida, D.V.; Bauer, A.; Fontaine, E.; Sordello, S.; Zaburannyi, N.; Herrmann, J.; Wenzel, S.C.; König, C. Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 2015, 348, 1106–1112. [Google Scholar] [CrossRef]
- Silver, L.L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Rizzi, M.; Rossi, F.; Miggiano, R. Mycobacterium tuberculosis UvrB forms dimers in solution and interacts with UvrA in the absence of ligands. Proteins 2018, 86, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, D.M.; Miggiano, R.; Rossi, F.; Rizzi, M. Mycobacterium tuberculosis Molecular Determinants of Infection, Survival Strategies, and Vulnerable Targets. Pathogens 2018, 7, 17. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grimwade, J.E.; Leonard, A.C. Blocking the Trigger: Inhibition of the Initiation of Bacterial Chromosome Replication as an Antimicrobial Strategy. Antibiotics 2019, 8, 111. https://doi.org/10.3390/antibiotics8030111
Grimwade JE, Leonard AC. Blocking the Trigger: Inhibition of the Initiation of Bacterial Chromosome Replication as an Antimicrobial Strategy. Antibiotics. 2019; 8(3):111. https://doi.org/10.3390/antibiotics8030111
Chicago/Turabian StyleGrimwade, Julia E., and Alan C. Leonard. 2019. "Blocking the Trigger: Inhibition of the Initiation of Bacterial Chromosome Replication as an Antimicrobial Strategy" Antibiotics 8, no. 3: 111. https://doi.org/10.3390/antibiotics8030111