Determination of Florfenicol, Thiamfenicol and Chloramfenicol at Trace Levels in Animal Feed by HPLC–MS/MS


 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. HPLC–MS/MS Conditions
2.2. Extraction
2.3. Method Validation
2.4. Real Sample Collection and Analysis
3. Materials and Methods
3.1. Chemicals, Reagents, and Stock Solutions
3.2. Conditions for HPLC–MS/MS Analysis
3.3. Sample Extraction
3.4. Method Validation
3.5. Sample Collection and Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- ESVAC (2015) Fifth ESVAC Report—Sales of Veterinary Antimicrobial Agents in 26 EU/EEA Countries in 2013. Available online: www.ema.europa.eu/docs/en_GB/document_library/Report/2015/10/WC500195687.pdf (accessed on 29 January 2017).
- Europe Comission. Regulation 37/2010. Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. Off. J. Eur. Union 2010, 15, 1–72. [Google Scholar]
- Europe Comission. Regulation 2019/4. Commission Regulation (EU) 2019/4 of the European Parliament and of the Council of 11 December 2018 on the manufacture, placing on the market and use of medicated feed, amending Regulation (EC) No 183/2005 of the European Parliament and of the Council and repealing Council Directive 90/167/EEC (Text with EEA relevance). Off. J. Eur. Union 2019, 4, 1–23. [Google Scholar]
- Samanidou, V.; Kaltzi, I.; Kabir, A.; Furton, K.G. Simplifying sample preparation using fabric phase sorptive extraction technique for the determination of benzodiazepines in blood serum by high performance liquid chromatography. Biomed. Chromatogr. 2016, 30, 829–836. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Wang, Y.; Pan, Y.; Tao, Y.; Chen, D.; Liu, Z.; Yuan, Z. Development and validation of a sensitive indirect competitive enzyme-linked immunosorbent assay for the screening of florfenicol and Tiamfenicol in edible animal tissue and feed. Food Anal. Method 2016, 9, 2434–2443. [Google Scholar] [CrossRef]
- Ge, S.; Yan, M.; Cheng, X.; Zhang, C.; Yu, J.; Zhao, P.; Gao, W. On-line molecular imprinted solid-phase extraction flow-injection fluorescence sensor for determination of florfenicol in animal tissues. J. Pharm. Biomed. 2010, 52, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J. Determination of florfenicol in fish feeds at high inclusion rates by HPLC-UV. J. AOAC Int. 2013, 96, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.; Konieczna, L.; Chmielewska, A.; Olędzka, I.; Plenis, A.; Bieniecki, M.; Lamparczyk, H. Comparative evaluation between capillary electrophoresis and high-performance liquid chromatography for the analysis of florfenicol in plasma. J. Pharm. Biomed. 2005, 39, 983–989. [Google Scholar] [CrossRef]
- Liu, H.Y.; Lin, S.L.; Fuh, M.R. Determination of chloramfenicol, Tiamfenicol and florfenicol in milk and honey using modified QuEChERS extraction coupled with polymeric monolith-based capillary liquid chromatography tandem mass spectrometry. Talanta 2016, 150, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Europe Comission. Comission Decission 2002/657/EC. Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Union 2002, 221, 8–36. [Google Scholar]
- Van de Riet, J.M.; Potter, R.A.; Christie-Fougere, M.; Burns, B.G. Simultaneous determination of residues of chloramfenicol, tiamfenicol, florfenicol, and florfenicol amine in farmed aquatic species by liquid chromatography/mass spectrometry. J. AOAC Int. 2003, 86, 510–514. [Google Scholar] [PubMed]
- Barreto, F.; Ribeiro, C.; Hoff, R.B.; Dalla Costa, T. Determination of chloramfenicol, Tiamfenicol, florfenicol and florfenicol amine in poultry, swine, bovine and fish by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2016, 1449, 48–53. [Google Scholar] [CrossRef]
- Anderson, S.C.; Subbiah, S.; Gentles, A.; Austin, G.; Stonum, P.; Brooks, T.A.; Smith, E.E. Qualitative and quantitative drug residue analyses: Florfenicol in white-tailed deer (Odocoileus virginianus) and supermarket meat by liquid chromatography tandem-mass spectrometry. J. Chromatogr. B 2016, 1033, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Wang, B.; Pang, M.; Zhao, X.; Xie, K.; Zhang, Y.; Dai, G. Quantitative analysis of chloramfenicol, tiamfenicol, florfenicol and florfenicol amine in eggs via liquid chromatography-electrospray ionization tandem mass spectrometry. Food Chem. 2018, 269, 542–548. [Google Scholar] [CrossRef]
- Pietro, W.J.; Woźniak, A.; Pasik, K.; Cybulski, W.; Krasucka, D. Amfenicols stability in medicated feed–development and validation of liquid chromatography method. Bull. Vet. Inst. Pulawy 2014, 58, 621–629. [Google Scholar] [CrossRef]
- Yang, J.; Sun, G.; Qian, M.; Huang, L.; Ke, X.; Yang, B. Development of a high-performance liquid chromatography method for the determination of florfenicol in animal feedstuffs. J. Chromatogr. B 2017, 1068, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Boix, C.; Ibáñez, M.; Sancho, J.V.; León, N.; Yusá, V.; Hernández, F. Qualitative screening of 116 veterinary drugs in feed by liquid chromatography–high resolution mass spectrometry: Potential application to quantitative analysis. Food Chem. 2014, 160, 313–320. [Google Scholar] [CrossRef]
- Robert, C.; Gillard, N.; Brasseur, P.Y.; Ralet, N.; Dubois, M.; Delahaut, P. Rapid multiresidue and multi-class screening for antibiotics and benzimidazoles in feed by ultra-high performance liquid chromatography coupled to tandem mass spectrometry. Food Control 2015, 50, 509–515. [Google Scholar] [CrossRef]
- Aguilera-Luiz, M.M.; Romero-González, R.; Plaza-Bolaños, P.; Vidal, J.L.M.; Frenich, A.G. Wide-scope analysis of veterinary drug and pesticide residues in animal feed by liquid chromatography coupled to quadrupole-time-of-flight mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 6543–6553. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Pérez, M.L.; Romero-González, R.; Martínez Vidal, J.L.; Garrido Frenich, A. Analysis of veterinary drug and pesticide residues in animal feed by high-resolution mass spectrometry: Comparison between time-of-flight and Orbitrap. Food Addit. Contam. A 2015, 32, 1637–1646. [Google Scholar] [CrossRef]
- León, N.; Pastor, A.; Yusà, V. Target analysis and retrospective screening of veterinary drugs, ergot alkaloids, plant toxins and other undesirable substances in feed using liquid chromatography–high resolution mass spectrometry. Talanta 2016, 149, 43–52. [Google Scholar] [CrossRef]
- Boscher, A.; Guignard, C.; Pellet, T.; Hoffmann, L.; Bohn, T. Development of a multi-class method for the quantification of veterinary drug residues in feedingstuffs by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 6394–6404. [Google Scholar] [CrossRef]
- Piatkowska, M.; Jedziniak, P.; Zmudzki, J. Multiresidue method for the simultaneous determination of veterinary medicinal products, feed additives and illegal dyes in eggs using liquid chromatography–tandem mass spectrometry. Food Chem. 2016, 197, 571–580. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Z.; Guo, X.; Cheng, L.; Wang, Z.; Shen, J. Simultaneous determination and confirmation of chloramfenicol, tiamfenicol, florfenicol and florfenicol amine in chicken muscle by liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2008, 875, 399–404. [Google Scholar] [CrossRef]
- Cronly, M.; Behan, P.; Foley, B.; Malone, E.; Martin, S.; Doyle, M.; Regan, L. Rapid multi-class multi-residue method for the confirmation of chloramfenicol and eleven nitroimidazoles in milk and honey by liquid chromatography-tandem mass spectrometry (LC-MS). Food Addit. Contam. 2010, 27, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, N.; Kojima, F.; Sugiura, K. Simultaneous determination of residues of chloramfenicol and florfenicol in animal feed by liquid chromatography tandem mass spectrometry. J. Resid. Sci. Technol. 2011, 8, 125–129. [Google Scholar]
- Wei, R.; Ge, F.; Chen, M.; Wang, R. Occurrence of ciprofloxacin, enrofloxacin, and florfenicol in animal wastewater and water resources. J. Environ. Qual. 2012, 41, 1481–1486. [Google Scholar] [CrossRef]
- Xiao, Z.; Song, R.; Rao, Z.; Wei, S.; Jia, Z.; Suo, D.; Fan, X. Development of a subcritical water extraction approach for trace analysis of chloramfenicol, tiamfenicol, florfenicol, and florfenicol amine in poultry tissues. J. Chromatogr. A 2015, 1418, 29–35. [Google Scholar] [CrossRef]
- Chou, K.Y.; Cheng, T.Y.; Chen, C.M.; Hung, P.L.; Tang, Y.Y.; Chung-Wang, Y.J.; Shih, Y.C. Simultaneous determination of residual tiamfenicol and florfenicol in foods of animal origin by HPLC/electrospray ionization-MS/MS. J. AOAC Int. 2009, 92, 1225–1232. [Google Scholar] [PubMed]
- Peng, L.I.; Yueming, Q.I.U.; Huixia, C.A.I.; Ying, K.O.N.G.; Yingzhang, T.A.N.G.; Daning, W.A.N.G.; Mengxia, X.I.E. Simultaneous determination of chloramfenicol, tiamfenicol, and florfenicol residues in animal tissues by gas chromatography/mass spectrometry. Chin. J. Chromatogr. 2006, 24, 14–18. [Google Scholar]
- Luo, P.; Chen, X.; Liang, C.; Kuang, H.; Lu, L.; Jiang, Z.; Shen, J. Simultaneous determination of thiamfenicol, florfenicol and florfenicol amine in swine muscle by liquid chromatography–tandem mass spectrometry with immunoaffinity chromatography clean-up. J. Chromatogr. B 2010, 878, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Nebot, C.; Iglesias, A.; Regal, P.; Miranda, J.; Cepeda, A.; Fente, C. Development of a multi-class method for the identification and quantification of residues of antibiotics, coccidiostats and corticosteroids in milk by liquid chromatography–tandem mass spectrometry. Int. Dairy J. 2012, 22, 78–85. [Google Scholar] [CrossRef]
- Nebot, C.; Guarddon, M.; Seco, F.; Iglesias, A.; Miranda, J.M.; Franco, C.M.; Cepeda, A. Monitoring the presence of residues of tetracyclines in baby food samples by HPLC-MS/MS. Food Control 2014, 46, 495–501. [Google Scholar] [CrossRef]
- Gavilán, R.; Nebot, C.; Miranda, J.; Martín-Gómez, Y.; Vázquez-Belda, B.; Franco, C.; Cepeda, A. Analysis of tetracyclines in medicated feed for food animal production by HPLC-MS/MS. Antibiotics 2016, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Gavilán, R.E.; Nebot, C.; Patyra, E.; Miranda, J.M.; Franco, C.M.; Cepeda, A. Simultaneous analysis of coccidiostats and sulphonamides in non-target feed by HPLC-MS/MS and validation following the Commission Decision 2002/657/EC. Food Addit. Contam. A 2018, 35, 1093–1106. [Google Scholar] [CrossRef] [PubMed]



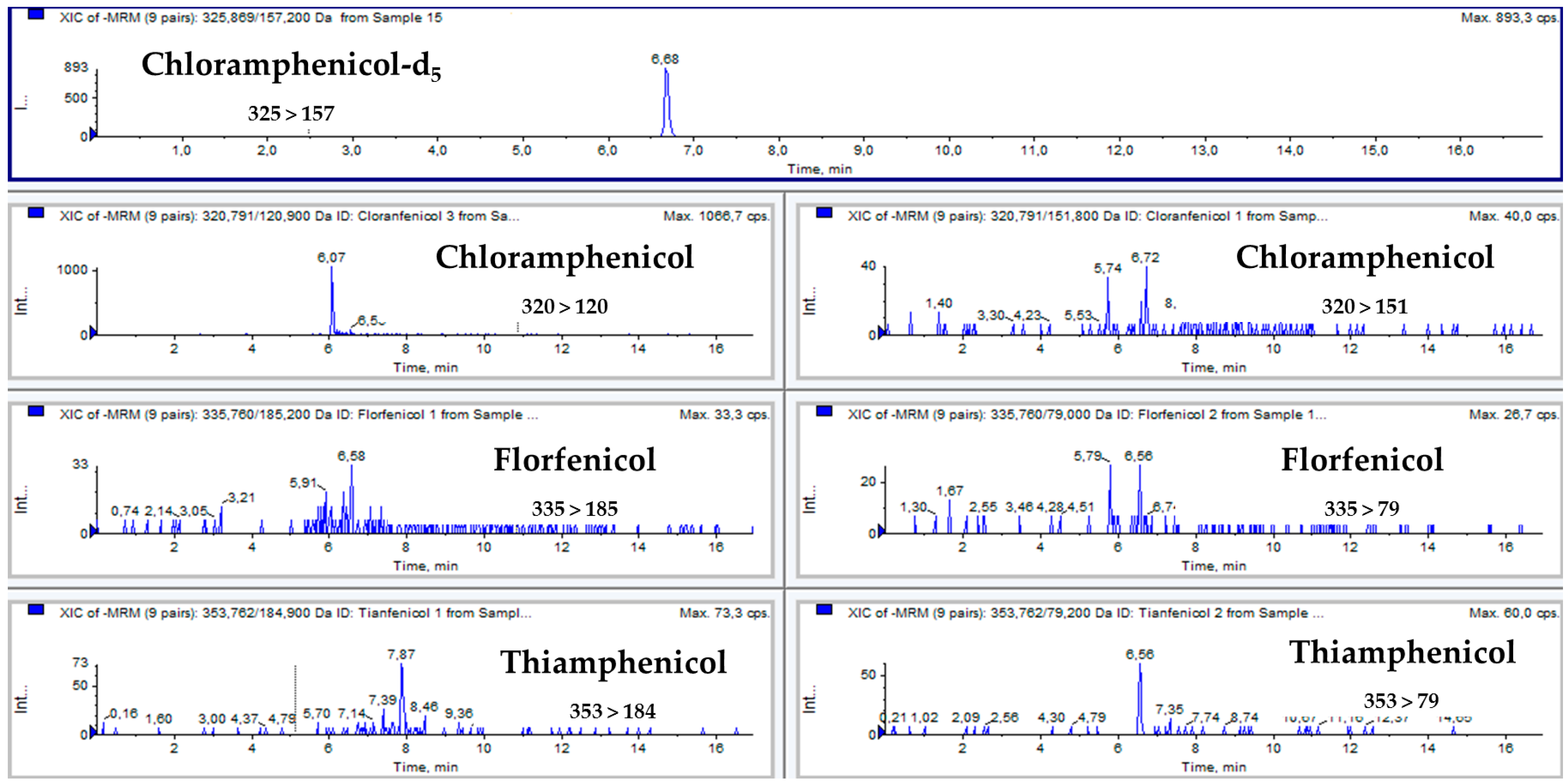{kind=link}
{kind=link}
| Tetracycline | Rt (min) | m/z Transition | CV | Collision Energy |
|---|---|---|---|---|
| Chloramfenicol | 6.71 | 320 > 151 | 46 | 26 |
| Chloramfenicol | 320 > 120 | 46 | 32 | |
| Thiamfenicol | 5.93 | 353 > 184 | 56 | 20 |
| Thiamfenicol | 353 > 79 | 56 | 27 | |
| Florfenicol | 6.57 | 335 > 185 | 61 | 23 |
| Florfenicol | 335 > 79 | 61 | 23 | |
| Chloramfenicol-d5 | 6.69 | 325 > 157 | 41 | 32 |
| Fortification Level (µg/kg) | Recovery (n = 18) | Repeatability (n = 6) | Reproducibility (n = 18) | CCα | CCβ | LOD | LOQ | |
|---|---|---|---|---|---|---|---|---|
| Chloramfenicol | 100 | 81 | 21 | 16 | 108 | 116 | 25 | 40 |
| 200 | 88 | 12 | 16 | |||||
| 300 | 89 | 15 | 19 | |||||
| Mean | 86 | 16 | 17 | |||||
| Tiamfenicol | 100 | 81.41 | 6.5 | 22.35 | 140 | 180 | 75 | 100 |
| 200 | 97.69 | 12.58 | 21.61 | |||||
| 300 | 103.83 | 18.97 | 21.98 | |||||
| Mean | 94.31 | 12.69 | 21.98 | |||||
| Florfenicol | 100 | 96.61 | 14.64 | 18.77 | 110 | 122 | 50 | 75 |
| 200 | 94.680 | 12.97 | 12.32 | |||||
| 300 | 90.342 | 13.22 | 15.22 | |||||
| Mean | 93.88 | 13.61 | 15.44 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavilán, R.E.; Nebot, C.; Patyra, E.; Vazquez, B.; Miranda, J.M.; Cepeda, A. Determination of Florfenicol, Thiamfenicol and Chloramfenicol at Trace Levels in Animal Feed by HPLC–MS/MS. Antibiotics 2019, 8, 59. https://doi.org/10.3390/antibiotics8020059
Gavilán RE, Nebot C, Patyra E, Vazquez B, Miranda JM, Cepeda A. Determination of Florfenicol, Thiamfenicol and Chloramfenicol at Trace Levels in Animal Feed by HPLC–MS/MS. Antibiotics. 2019; 8(2):59. https://doi.org/10.3390/antibiotics8020059
Chicago/Turabian StyleGavilán, Rosa Elvira, Carolina Nebot, Ewelina Patyra, Beatriz Vazquez, Jose Manuel Miranda, and Alberto Cepeda. 2019. "Determination of Florfenicol, Thiamfenicol and Chloramfenicol at Trace Levels in Animal Feed by HPLC–MS/MS" Antibiotics 8, no. 2: 59. https://doi.org/10.3390/antibiotics8020059