
Chloramphenicol Derivatives as Antibacterial and Anticancer Agents: Historic Problems and Current Solutions

 ,
,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
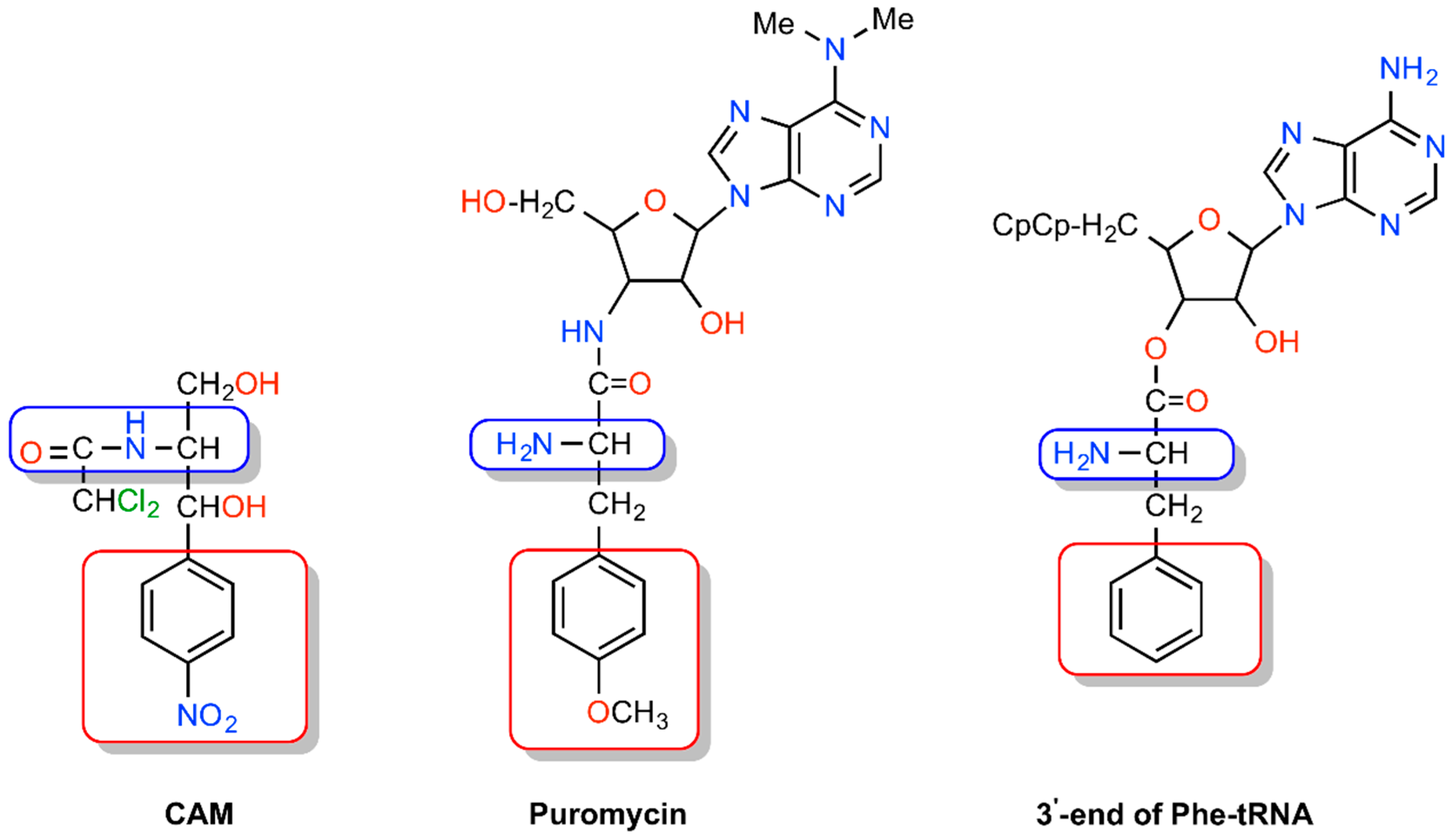{kind=link}
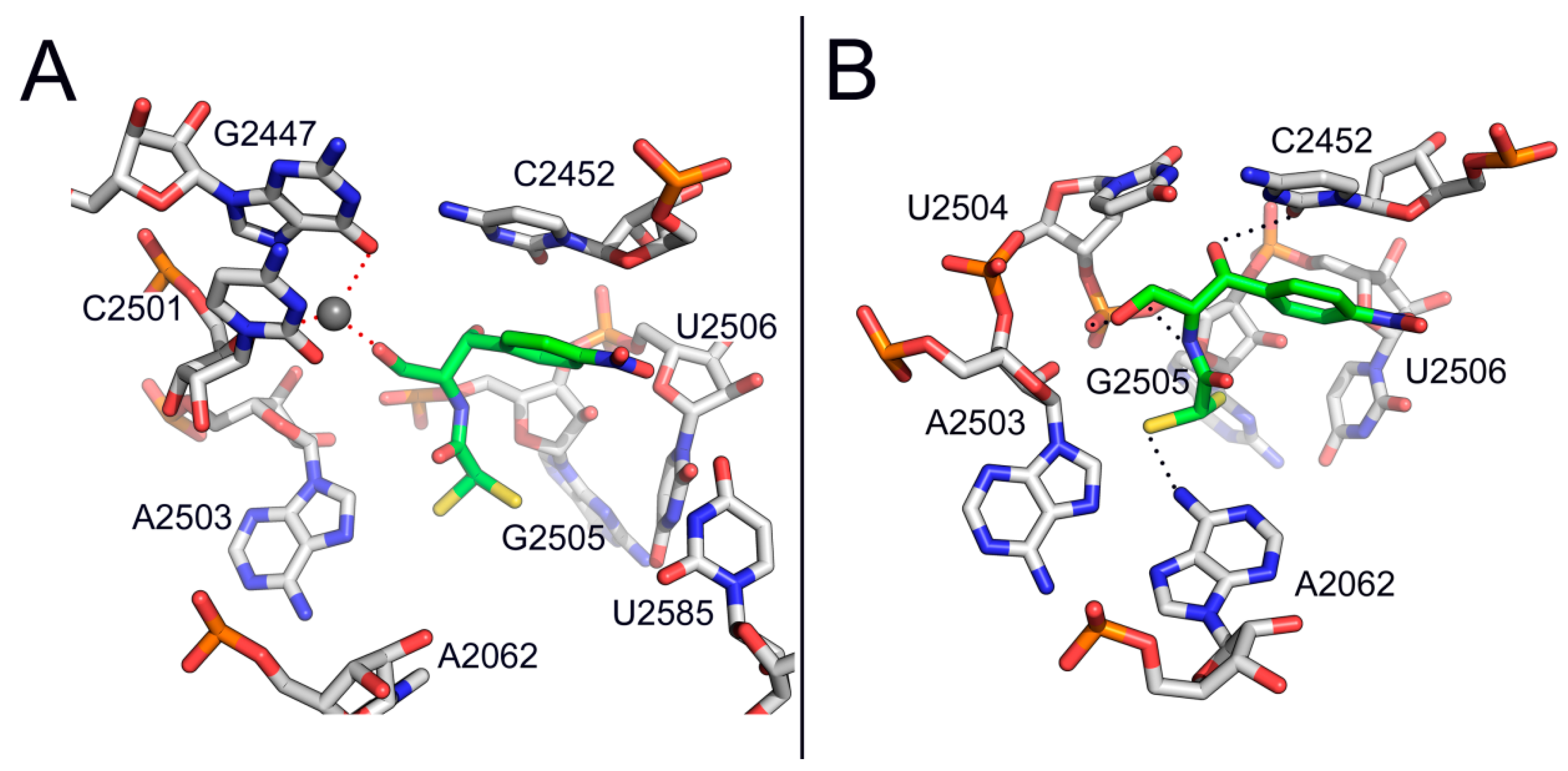{kind=link}
{kind=link}
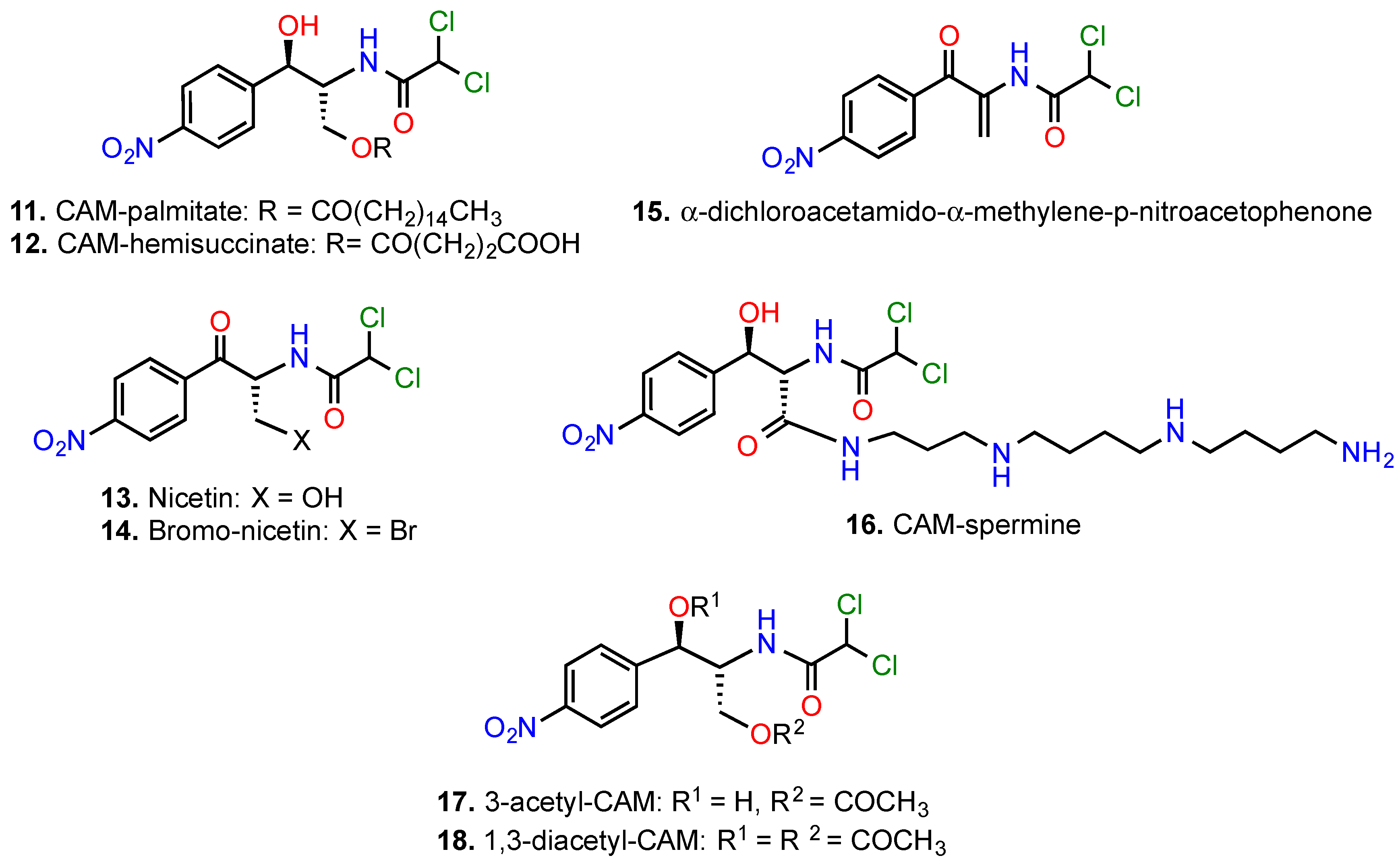{kind=link}
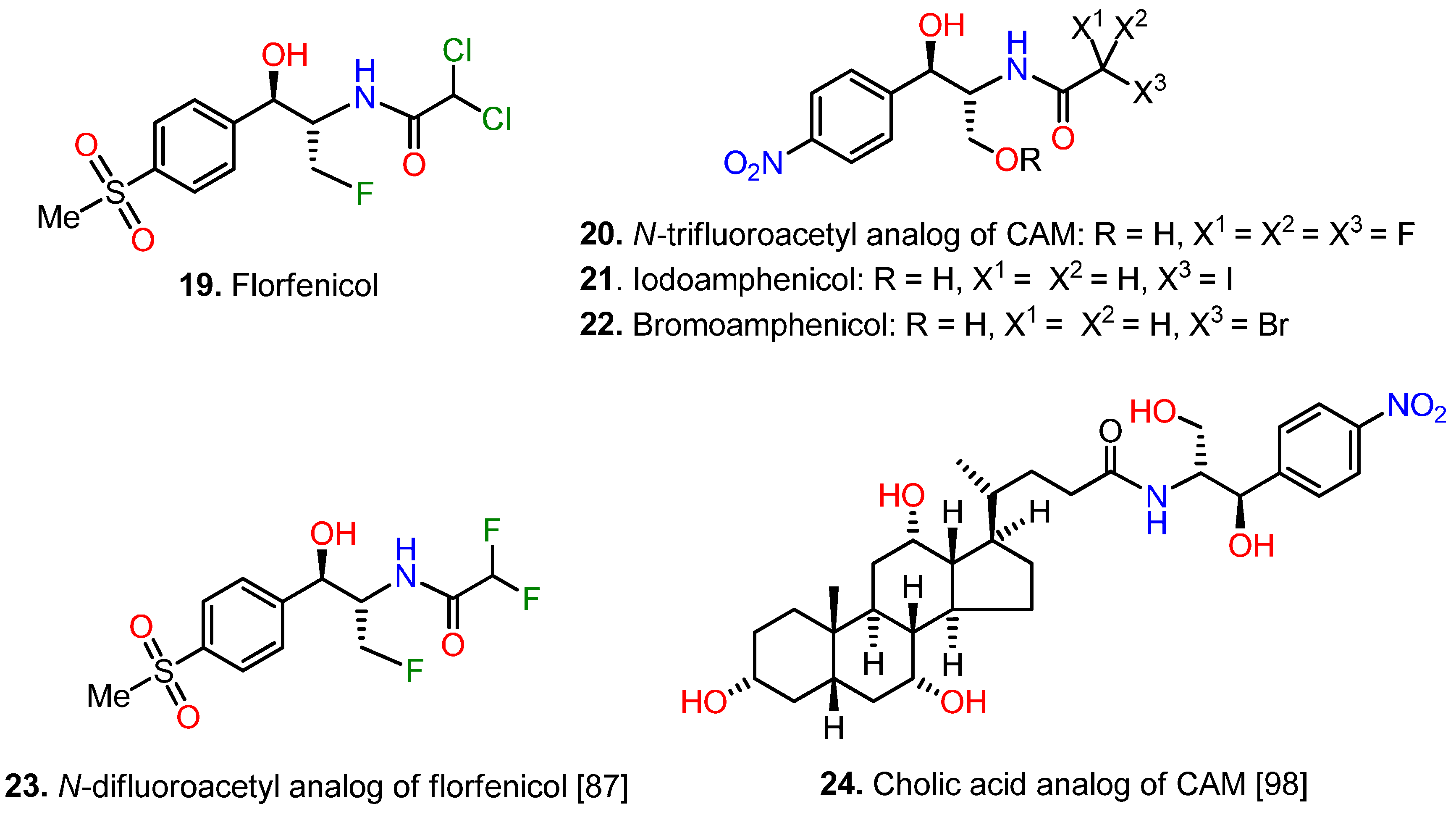{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
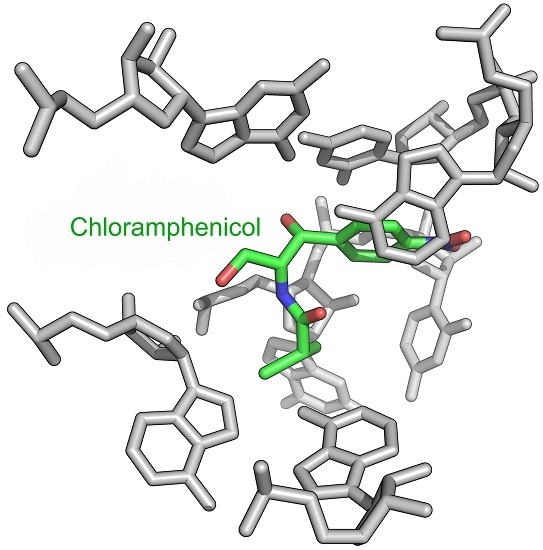
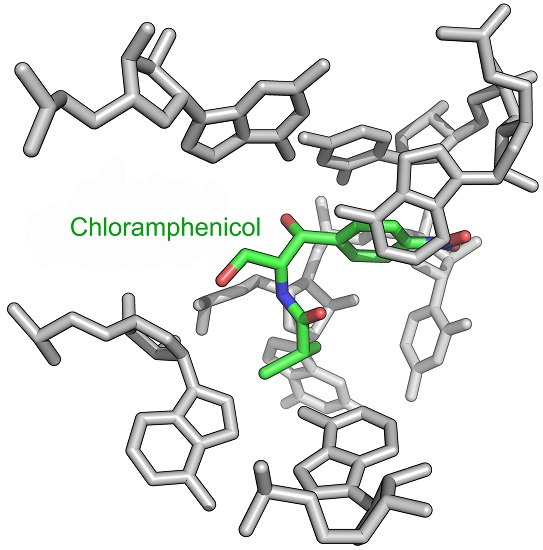
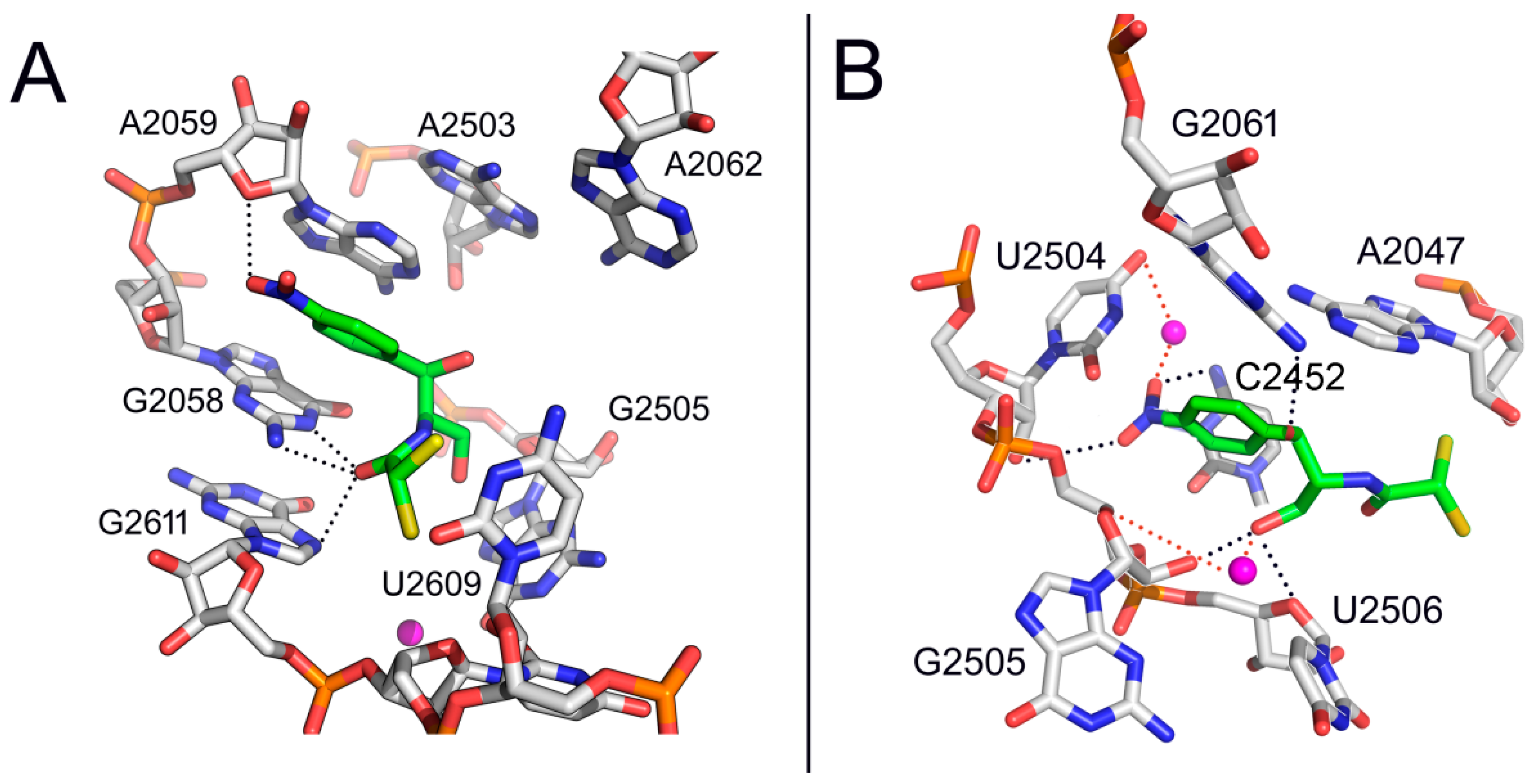
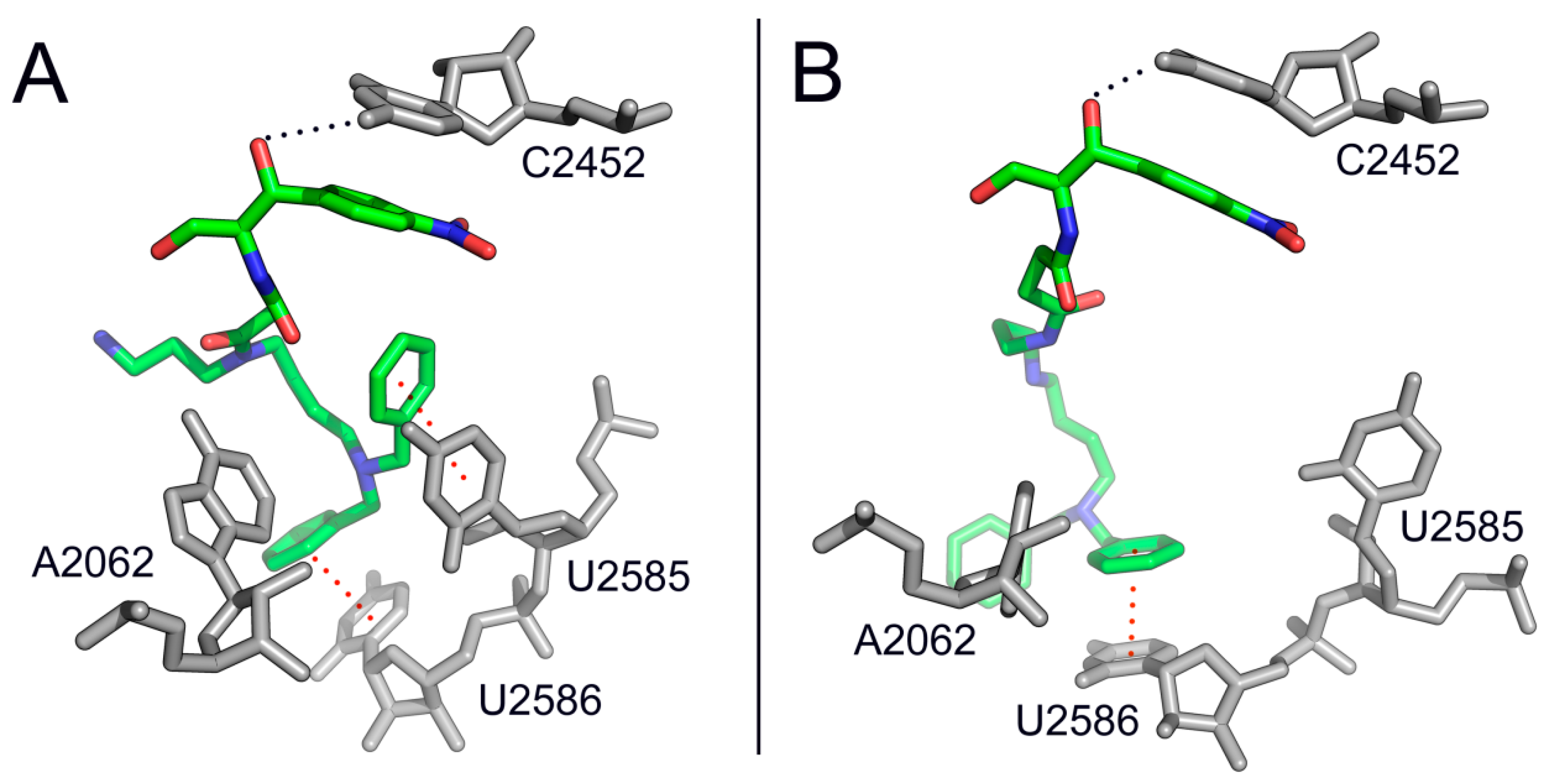
2. Mode of Action of CAM
3. Bacterial Survival Strategies to Combat CAM Activity
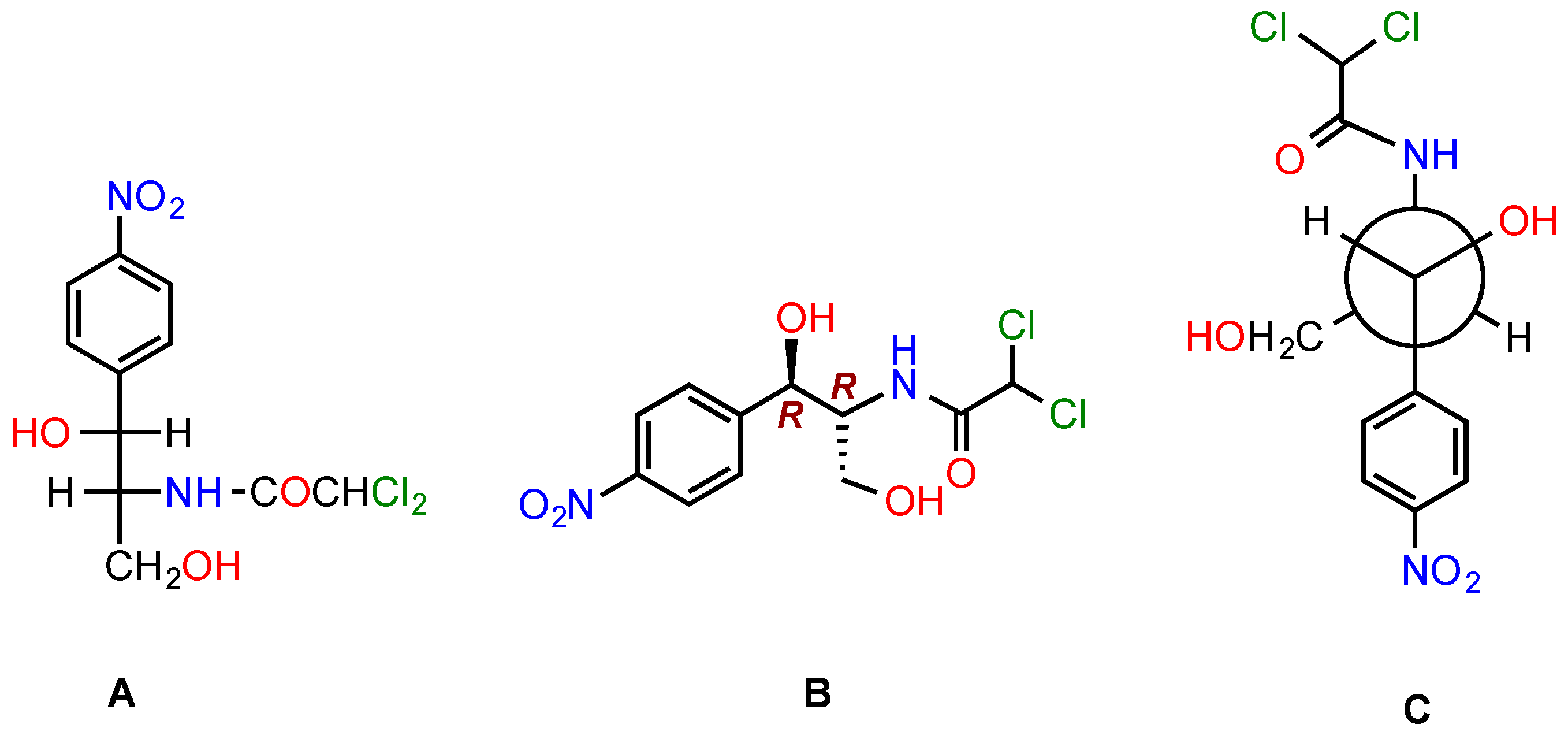
4. Side Effects of CAM
5. Modifications of CAM to Obtain Antibacterials with Improved Properties
5.1. Modifications of the p-Nitrophenyl Moiety
5.2. Modifications of the 2-Amino-1,3-propanediol Moiety
5.3. Modifications at Both the p-Nitrophenyl and the 2-Amino-1,3-propanediol Moieties
5.4. Modifications at the Dichloroacetyl Moiety
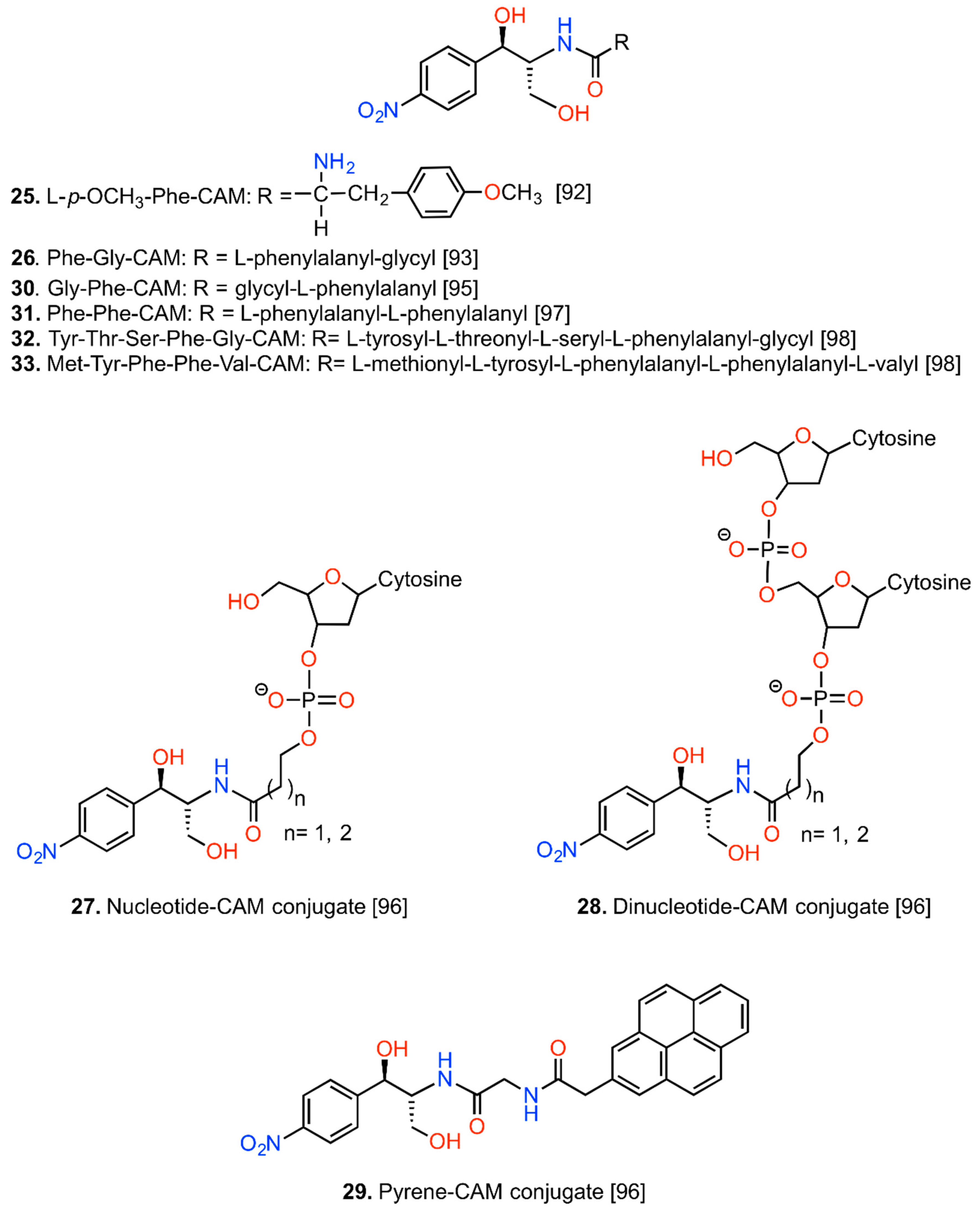
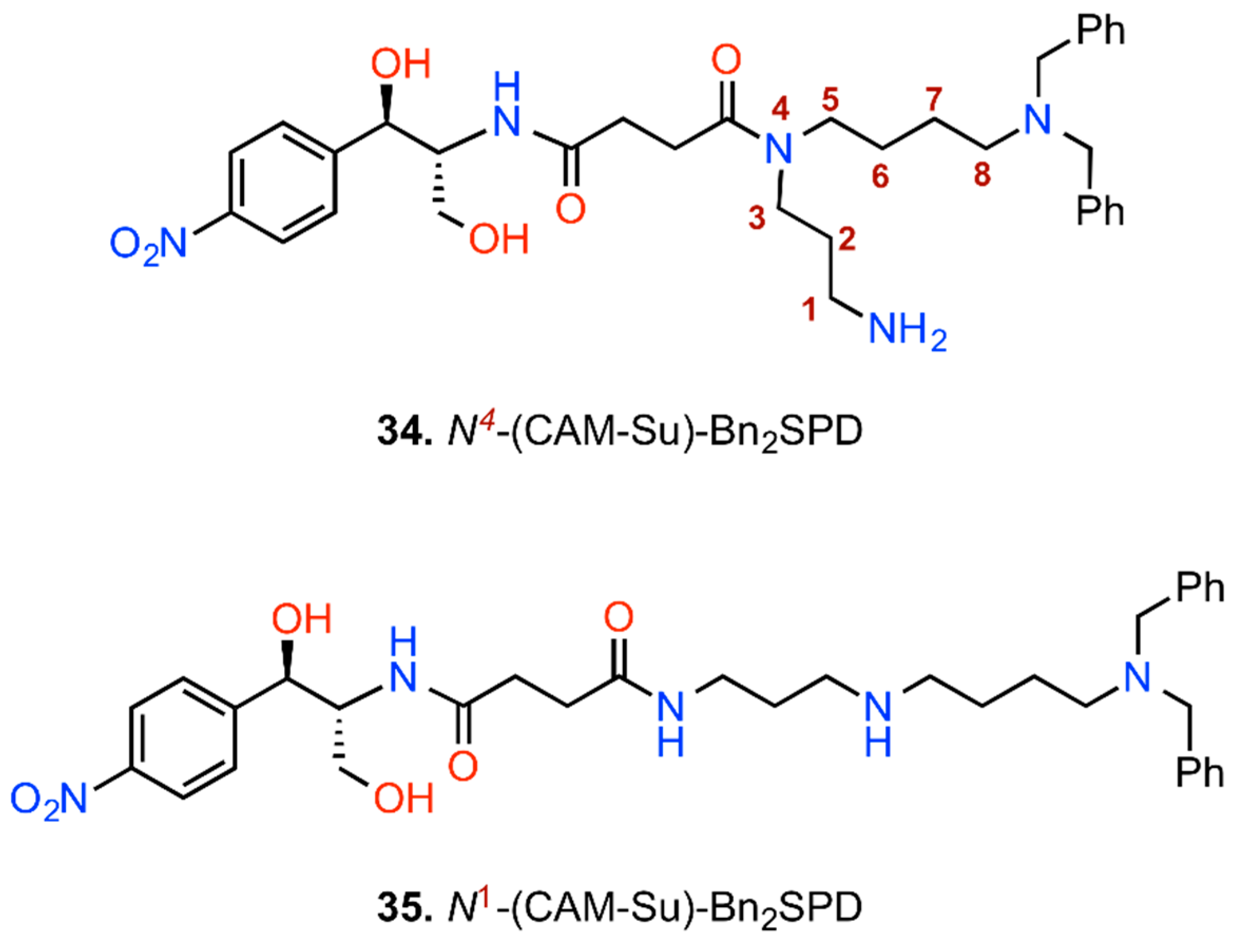
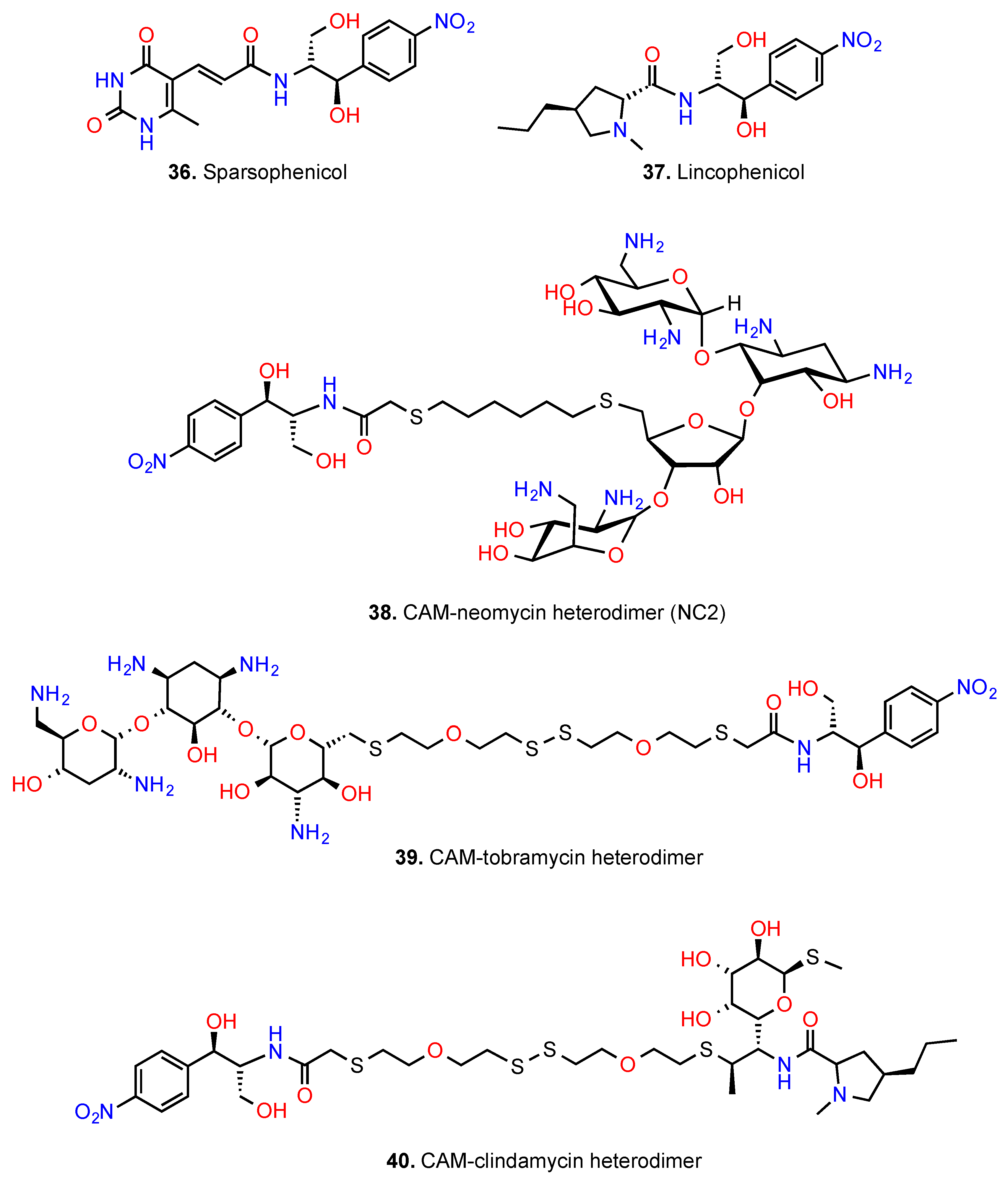
6. CAM Hybrids and Dimers
6.1. CAM Hybrids
6.2. CAM Heterodimers
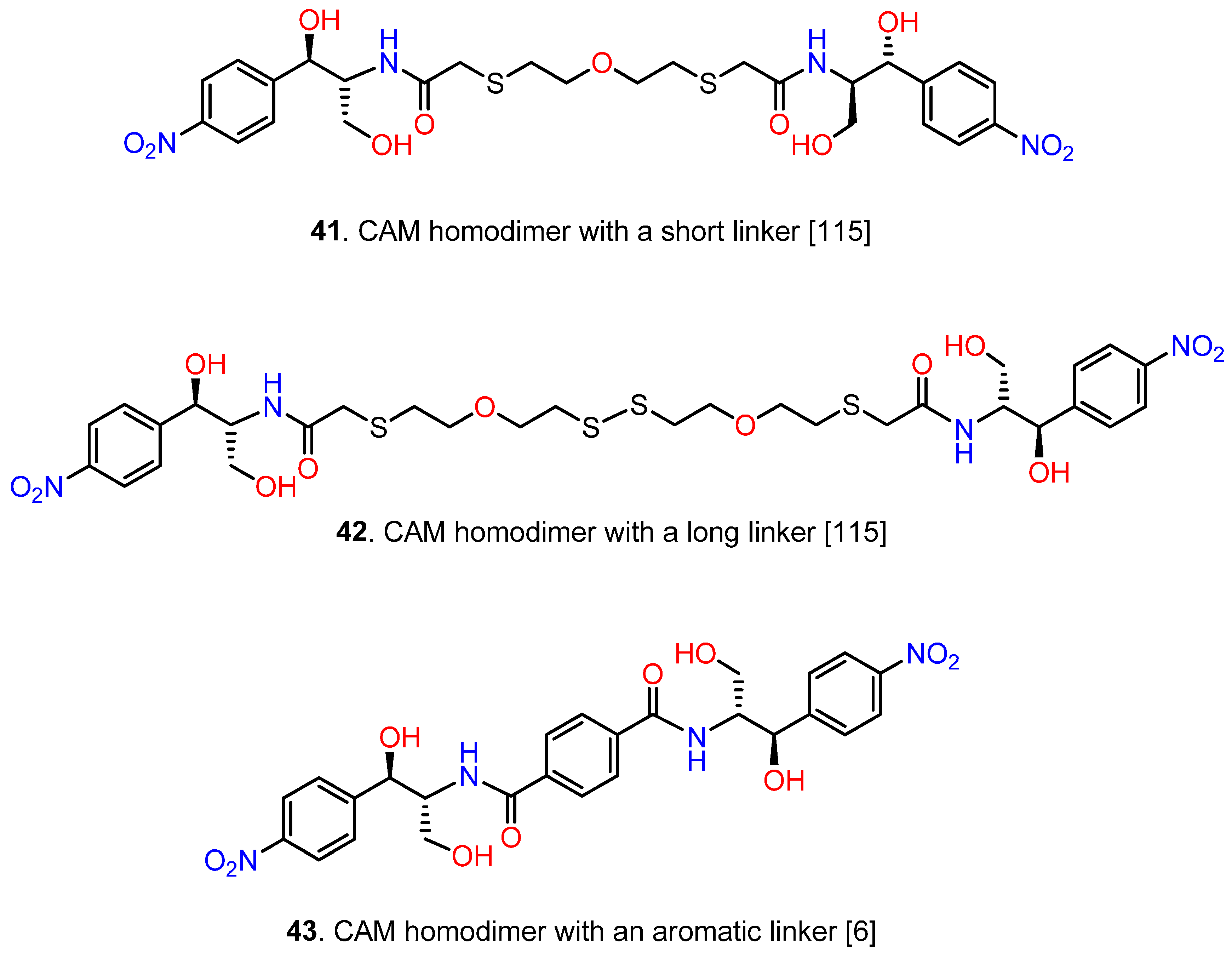
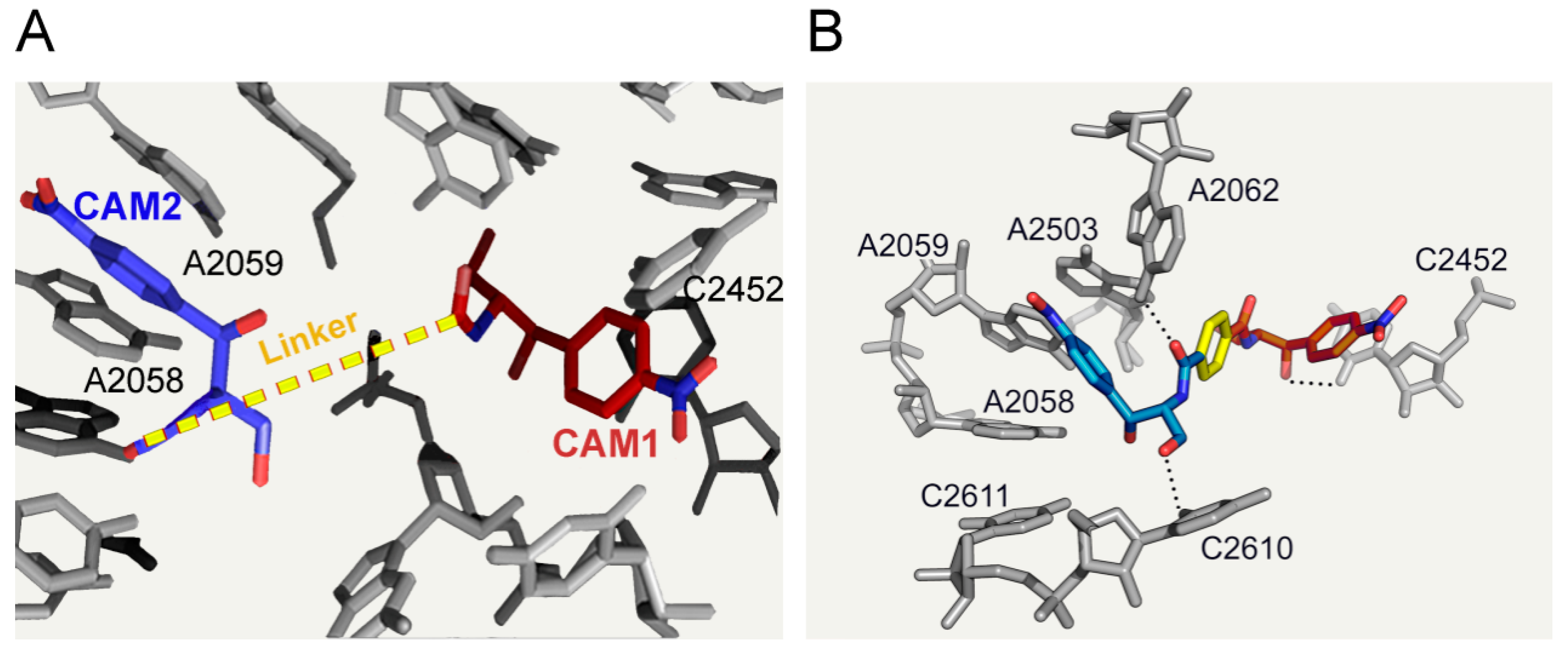
6.3. CAM Homodimers
7. Synopsis and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ehrlich, J.; Bartz, Q.R.; Smith, R.M.; Joslyn, D.A.; Burkholder, P.R. Chloromycetin, a new antibiotic from a soil actinomycete. Science 1947, 106, 417. [Google Scholar] [CrossRef] [PubMed]
- Pongs, O. Chloramphenicol. In Mechanism of Action of Antibacterial Agents; Hann, F.E., Ed.; Springer: New York, NY, USA, 1979; Volume 5, pp. 26–42. [Google Scholar]
- Contreras, A.; Vazquez, D. Cooperative and antagonistic interactions of peptidyl-tRNA and antibiotics with bacterial ribosomes. Eur. J. Biochem. 1977, 74, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Long, K.S.; Porse, B.T. A conserved chloramphenicol binding site at the entrance to the ribosomal peptide exit tunnel. Nucleic Acids Res. 2003, 31, 7208–7215. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.L.; Moore, P.B.; Steitz, T.A. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 2003, 330, 1061–1075. [Google Scholar] [CrossRef]
- Kostopoulou, O.N.; Magoulas, G.E.; Papadopoulos, G.E.; Mouzaki, A.; Dinos, G.P.; Papaioannou, D.; Kalpaxis, D.L. Synthesis and evaluation of chloramphenicol homodimers: Molecular target, antimicrobial activity, and toxicity against human cells. PLoS ONE 2015, 10, e0134526. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Le Mahieu, R.A. Inhibition of [14C] chloramphenicol binding to Escherichia coli ribosomes by erythromycin derivatives. Antimicrob. Agents Chemother. 1974, 6, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Rheinberger, H.J.; Nierhaus, K.H. Partial release of AcPhe-transfer RNA from ribosomes during poly(U)-dependent poly(Phe) synthesis and the effects of chloramphenicol. Eur. J. Biochem. 1990, 193, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Vester, B.; Garrett, R.A. The importance of highly conserved nucleotides in the binding region of chloramphenicol at the peptidyl transferase center of Escherichia coli 23S ribosomal RNA. EMBO J. 1988, 7, 3577–3587. [Google Scholar] [PubMed]
- Douthwaite, S. Functional interactions within 23S rRNA involving the peptidyltransferase center. J. Bacteriol. 1992, 174, 1333–1338. [Google Scholar] [PubMed]
- Giessing, A.M.; Jensen, S.S.; Rasmussen, A.; Hansen, L.H.; Gondela, A.; Long, K.; Vester, B.; Kirpekar, F. Identification of 8-methyladenosine as the modification catalyzed by the radical SAM methyltransferase Cfr that confers antibiotic resistance in bacteria. RNA 2009, 15, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Persaud, C.; Lu, Y.; Vila-Sanjurjo, A.; Campell, J.L.; Finley, J.; O’Connor, M. Mutagenesis of the modified bases, m5U1939 and ψ2504, in Escherichia coli 23S rRNA. Biochem. Biophys. Res. Commun. 2010, 392, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Celma, M.L.; Monro, R.E.; Vazquez, D. Substrate and antibiotic binding sites at the peptidyl transferase center of E. coli ribosomes: Binding of UACCA-leu to 50S subunits. FEBS Lett. 1971, 13, 247–251. [Google Scholar] [CrossRef]
- Fernandez-Muñoz, R.; Vazquez, D. Kinetic studies of peptide bond formation. Effect of chloramphenicol. Mol. Biol. Rep. 1973, 1, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S. The use of inhibitors in studies on protein synthesis. Methods Enzymol. 1974, 30, 261–282. [Google Scholar] [PubMed]
- Drainas, D.; Kalpaxis, D.L.; Coutsogeorgopoulos, C. Inhibition of ribosomal peptidyl transferase by chloramphenicol: Kinetic studies. Eur. J. Biochem. 1987, 164, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Xaplanteri, M.; Andreou, A.; Dinos, G.P.; Kalpaxis, D.L. Effect of polyamines in the inhibition of peptidyl transferase by antibiotics: Revisiting the mechanism of chloramphenicol action. Nucleic Acids Res. 2003, 31, 5074–5083. [Google Scholar] [CrossRef] [PubMed]
- Schlünzen, F.; Zarivach, R.; Harms, J.; Bashan, A.; Tocilj, A.; Albrecht, R.; Yonath, A.; Franceschi, F. Structural basis for the interaction of antibiotics with the peptidyl transferase center in eubacteria. Nature 2001, 413, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Dunkle, J.A.; Xiong, L.; Mankin, A.S.; Cate, J.H.D. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. USA 2010, 107, 17152–17157. [Google Scholar] [CrossRef] [PubMed]
- Bulkley, D.; Innis, C.A.; Blaha, G.; Steitz, T.A. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc. Natl. Acad. Sci. USA 2010, 107, 17158–17163. [Google Scholar] [CrossRef] [PubMed]
- Kirillov, S.V.; Porse, B.T.; Garrett, R.A. Peptidyl transferase antibiotics perturb the relative positioning of the 3′-terminal adenosine of P/P′-site-bound tRNA and 23S rRNA in the ribosome. RNA 1999, 5, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Cheney, B.V. Ab initio calculations on the large molecules using molecular fragments. Structural correlations between natural moieties and some antibiotic inhibitors of peptidyl transferase. J. Med. Chem. 1974, 17, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Bhuta, P.; Chung, H.L.; Hwang, J.-S.; Žemlička, J. Analogues of chloramphenicol: Circular dichroism spectra, inhibition of ribosomal peptidyl tranferase, and possible mechanism of action. J. Med. Chem. 1980, 23, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.; Chorev, M. On the concept of linear modified retro-peptide structures. Acc. Chem. Res. 1979, 12, 1–7. [Google Scholar] [CrossRef]
- Monro, R.E.; Staehelin, T.; Celma, M.L.; Vazquez, D. The peptidyl transferase activity of ribosomes. Cold Spring Harb. Symp. Quant. Biol. 1969, 34, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S. Studies on the formation of transfer ribonucleic acid-ribosome complexes. 8. Survey of the effect of antibiotics of N-acetyl-phenylalanine-puromycine formation: Possible mechanism of chloramphenicol action. Arch. Biochem. Biophys. 1970, 136, 80–88. [Google Scholar] [CrossRef]
- Kalpaxis, D.L.; Coutsogeorgopoulos, C. Type of inhibition of peptide bond formation by chloramphenicol depends on the temperature and the concentration of ammonium ions. Mol. Pharmacol. 1989, 36, 615–619. [Google Scholar] [PubMed]
- Morrison, J.B.; Walsh, C.T. The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 1988, 61, 201–301. [Google Scholar] [PubMed]
- Pestka, S. Studies on the formation of transfer ribonucleic acid-ribosome complexes. XI. Antibiotic effects on phenylalanine-oligonucleotide binding to ribosomes. Proc. Natl. Acad. Sci. USA 1969, 64, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Polacek, N.; Gomez, M.J.; Ito, K.; Xiong, L.; Nakamura, Y.; Mankin, A.S. The critical role of universally conserved A2602 of 23S ribosomal RNA in the release of the nascent peptide during translation termination. Mol. Cell 2003, 11, 103–112. [Google Scholar] [CrossRef]
- Thompson, J.; O’Connor, M.; Mills, J.A.; Dahlberg, A.E. The protein synthesis inhibitors, oxazolidinones and chloramphenicol, cause extensive translational inaccuracy in vivo. J. Mol. Biol. 2002, 322, 273–279. [Google Scholar] [CrossRef]
- Champey, W.S. The other target for ribosomal antibiotics inhibition of bacterial subunit formation. Infect. Disord. Drug Targets 2006, 6, 377–390. [Google Scholar] [CrossRef]
- Silback, T.; Peil, L.; Xiong, L.; Mankin, A.; Remme, J.; Tenson, T. Erythromycin- and chloramphenicol-induced ribosomal assembly defects are secondary effects of protein synthesis inhibition. Antimicrob. Agents Chemother. 2009, 53, 563–571. [Google Scholar]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Osawa, S.; Takata, R. Chloramphenicol resistant mutants of Bacillus subtilis. Mol. Gen. Genet. 1973, 127, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Shen, Z.; Naren, G.; Li, H.; Xia, X.; Wu, C.; Shen, J.; Zhang, Q.; Wang, Y. Identification of a novel G2073A mutation in 23S rRNA in amphenicol-selected mutants of Campylobacter jejuni. PLoS ONE 2014, 9, e94503. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Kehrenberg, C.; Doublet, B.; Cloeckaert, A. Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol. Rev. 2004, 28, 519–542. [Google Scholar] [CrossRef] [PubMed]
- Mosher, R.H.; Camp, D.J.; Yang, K.; Brown, M.P.; Shaw, W.W.; Vining, L.C. Inactivation of chloramphenicol by O-phosphorylation. J. Biol. Chem. 1995, 27, 27000–27006. [Google Scholar] [CrossRef]
- Izard, T.; Ellis, J. The crystal structure of chloramphenicol phosphotransferase reveals a novel inactivation mechanism. EMBO J. 2000, 19, 2690–2700. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, T.; Sung, C.; Kim, H.; Song, E.; Park, H.Y.; Jeon, J.M.; Yoo, D.; Kim, H.J.; Kim, Y.H.; Choi, K.Y.; et al. Phosphorylation of chloramphenicol by a recombinant protein Yhr2 from Streptomyces avermitilis MA4680. Bioorg. Med. Chem. Lett. 2013, 23, 3614–3619. [Google Scholar] [CrossRef] [PubMed]
- Mosher, R.H.; Ranade, N.P.; Schrempf, H.; Vining, L.C. Chloramphenicol resistance in Streptomyces, cloning and characterization of a chloramphenicol hydrolase gene from Streptomyces venezuelae. J. Gen. Microbiol. 1990, 136, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Tao, W; Lee, M.H.; Wu, J.; Kim, N.H.; Kim, J.-C.; Chung, E.; Hwang, E.C.; Lee, S.-W. Inactivation of chloramphenicol and florfenicol by a novel chloramphenicol hydrolase. Appl. Environ. Microbiol. 2012, 28, 6295–6301. [Google Scholar]
- Smith, A.L.; Erwin, A.L.; Kline, T.; Unrath, W.C.; Nelson, K.; Weber, A.; Howard, W.N. Chloramphenicol is a substrate for a novel nitroreductase pathway in Heamophila influenzae. Antimicrob. Agents Chemother. 2007, 51, 2820–2829. [Google Scholar] [CrossRef] [PubMed]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 2009, 1794, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Danilchanka, O.; Pavienok, M.; Niederweis, M. Role of porins for uptake of antibiotics by Mycobacterium smegmatis. Antimicrob. Agents Chemother. 2008, 52, 3127–3134. [Google Scholar] [CrossRef] [PubMed]
- Kadlec, K.; Kerhrenberg, C.; Schwarz, S. Efflux-mediated resistance to florfenicol and/or chloramphenicol in Bordetella bronchiseptica: Identification of a novel chloramphenicol exporter. J. Antimicrob. Chemoth. 2007, 59, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Y.; Wu, C.; Schwarz, S.; Shen, Z.; Jeon, B.; Ding, S.; Zhang, Q.; Shen, J. A novel phenicol exporter gene, fexB, found in enterococci of animal origin. J. Antimicrob. Chemother. 2012, 67, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Fluman, N.; Bibi, E. Bacterial multidrug transport through the lens of the major facilitator superfamily. Biochim. Biophys. Acta 2009, 1794, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Bay, D.C.; Rommens, K.L.; Turner, R.J. Small multidrug resistance proteins: A multidrug transporter family that continues to grow. Biochim. Biophys. Acta 2008, 1778, 1814–1838. [Google Scholar] [CrossRef] [PubMed]
- Magnet, S.; Courvalin, P.; Lambert, T. Resistance-nodulation-cell division-type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454. Antimicrob. Agents Chemother. 2001, 45, 3375–3380. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.M.; George, A.M. The ABC transporter structure and mechanism: Perspectives on recent research. Cell. Mol. Life Sci. 2004, 61, 682–699. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, T.; Tsuchiya, T. Multidrug efflux transporters in the MATE family. Biochim. Biophys. Acta 2009, 1794, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, K.; Sobel, M.L.; Garch, F.F.; Poole, K.; Plesiat, P. Induction of the MexXY efflux pump in Pseudomonas aeruginosa is dependent on drug-ribosome interaction. J. Bacteriol. 2005, 187, 5341–5346. [Google Scholar] [CrossRef] [PubMed]
- Kinama, A.D.; Yargiu, A.Y.; Nikaido, H. Some ligands enhance the efflux of other ligands by the Escherichia coli multi drug pump AcrB. Biochemistry 2013, 52, 8342–8351. [Google Scholar] [CrossRef] [PubMed]
- Ersler, A.J.; Iossifides, I.A. In vitro action of chloramphenicol-analogues on the metabolism of human red blood cells. Αcta Haemat. 1962, 28, 1–19. [Google Scholar] [CrossRef]
- Yunis, A.A.; Arimura, G.K.; Isildar, M. DNA damage induced by chloramphenicol and its nitroso derivative: Damage in intact cells. Am. J. Hematol. 1987, 24, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, D.; Dabrowska, M.; Jakoniuk, P.; Rózański, A. Pyrrole analogues of chloramphenicol. I. Synthesis and antibacterial activity of DL-threo-1-(l-methyl-4-nitro-pyrrole-2-yl)-2-dichloroacetamidopropane-1,3-diol. Acta Pol. Pharm. 2000, 57, 213–221. [Google Scholar] [PubMed]
- Ohnisi, S.; Murata, M.; Ida, N.; Oikawa, S.; Kawanishi, S. Oxidative DNA damage induced by metabolites of chloramphenicol, an antibiotic drug. Free Radic. Res. 2015, 49, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Yunnis, A.A. Chloramphenicol-induced bone marrow suppression. Semin. Hematol. 1973, 10, 225–234. [Google Scholar]
- Jones, C.N.; Miller, C.; Tenenbaum, A.; Spremmuli, L.L.; Saada, A. Antibiotic effects on mitochondrial translation and in patients with mitochondrial translation defects. Mitochondrion 2009, 9, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Amunts, A.; Brown, A. Organization and regulation of mitochondrial protein synthesis. Annu. Rev. Biochem. 2016, 85. [Google Scholar] [CrossRef] [PubMed]
- Turton, J.A.; Fagg, R.; Sones, W.R.; Williams, T.C.; Andrews, C.M. Characterization of the myelotoxicity of chloramphenicol succinate in the B6C3F1 mouse. Int. J. Exp. Pathol. 2006, 87, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Nau, R.; Sörgel, F.; Eiffert, H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin. Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef] [PubMed]
- Musa, S.H.; Basri, M.; Masoumi, H.R.; Karjiban, R.A.; Malek, E.A.; Basri, H.; Shamsuddin, A.F. Formulation optimization of palm kernel oil esters nanoemulsion-loaded with chloramphenicol suitable for meningitis treatment. Colloids Surf. B Biointerfaces 2013, 112, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Sadum, A.A. Drug-related mitochondrial optic neuropathies. J. Neuroophthalmol. 2013, 33, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Sachs, B.; Erdmann, T.; Al Masaoudi, T.; Merk, H.F. Molecular features determining hymphocyte reactivity in allergic contact dermatitis to chloramphenicol and azidamphenicol. Allergy 2001, 56, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Shuang, G.; Yu, S.; Guo, W.; Wang, D.; Zhang, Z.; Lu, J.; Deng, X. Immunosupressive activity of florfenicol on the immune responses in mice. Immunol. Invest. 2011, 40, 356–366. [Google Scholar] [CrossRef] [PubMed]
- McIntype, J.; Choonara, I. Drug toxicity in the neonate. Biol. Neonate 2004, 86, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Cheng, Y.W.; Liao, P.L.; Yang, Y.T.; Kang, J.J. Chloramphenicol causes mitochondrial stress, decreases ATP biosynthesis, induces matrix metalloproteinase-13 expression, and solid-tumor invasion. Toxicol. Sci. 2010, 116, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [PubMed]
- Lokhande, J.; Juvekar, A.S.; Kulkarni, K.P. Chloramphenicol: Screening and review to evaluate the potential beneficial effects in leukaemia. J. Indian Med. Assoc. 2007, 105, 224–228. [Google Scholar] [PubMed]
- Kostopoulou, O.N.; Kouvela, E.C.; Magoulas, G.E.; Garnelis, T.; Panagoulias, I.; Rodi, M.; Papadopoulos, G.; Mouzaki, A.; Dinos, G.P.; Papaioannou, D.; et al. Conjugation with polyamines enhances the antibacterial and anticancer activity of chloramphenicol. Nucleic Acids Res. 2014, 42, 8621–8634. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.R.; Shi, Y. Chloramphenicol induces abnormal differentiation and inhibits apoptosis in activated T cells. Cancer Res. 2008, 68, 4875–4881. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, G.; Gross, H.; Bladler, G. Synthesis and antibacterial activity of perchlorylchloramphenicol. Pharmazie 1983, 38, 587–589. [Google Scholar] [PubMed]
- Yunis, A.A.; Manyan, D.R.; Arimura, G.K. Comparative effect of chloramphenicol and thiamphenicol on DNA and mitochondrial protein synthesis in mammalian cells. J. Lab. Clin. Med. 1973, 81, 713–718. [Google Scholar] [PubMed]
- Mullen, G.B.; Georgiev, V.S. Adamantylmethyl analogues of chloramphenicol. J. Pharm. Sci. 1988, 77, 643–644. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.D.; Gerry, C.J.; Tan, D.S. General platform for systematic quantitative evaluation of small molecule permeability in bacteria. ACS Chem. Biol. 2014, 9, 2535–2544. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, D.; Dabrowska, M.; Jakoniuk, P.; Rózański, A. Pyrrole analogues of chloramphenicol. III. Synthesis and antibacterial activity of DL-threo-1-(1-methyl-sulfonyl]pyrrole-3-yl)-2-dichlorocyclopropane-1,3-diol. Acta Pol. Pharm. 2002, 59, 127–132. [Google Scholar] [PubMed]
- Ambrose, P.J. Clinical pharmacokinetics of chloramphenicol and chloramphenicol succinate. Clin. Pharmacokinet. 1984, 9, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Brady, A.J.; Gray, J.D. The treatment of fungous infection of the skin with nicetin. Can. Med. Assoc. J. 1957, 76, 725–729. [Google Scholar] [PubMed]
- Kono, M.; O’Hara, K.; Honda, M.; Mitsuhashi, S. Drug resistance of Staphylococci. XI. Induction of chloramphenicol resistance by its derivatives and analogues. J. Antibiot. (Tokyo) 1969, 22, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Hahn, F.E. New derivatives of chloramphenicol active against resistant bacteria. Naturwissenschaften 1980, 67, 89–90. [Google Scholar] [CrossRef] [PubMed]
- Magoulas, G.E.; Kostopoulou, O.N.; Garnelis, T.; Athanassopoulos, C.M.; Kournoutou, G.G.; Leotsinidis, M.; Dinos, G.P.; Papaioannou, D.; Kalpaxis, D.L. Synthesis and antimicrobial activity of chloramphenicol-polyamine conjugates. Bioorg. Med. Chem. 2015, 23, 3163–3174. [Google Scholar] [CrossRef] [PubMed]
- Piffaretti, J.C.; Allet, B.; Pitton, J.S. Analogy between in vivo and in vitro biological effect of chloramphenicol and its acetylated derivatives. FEBS Lett. 1970, 11, 26–28. [Google Scholar] [CrossRef]
- Neu, H.C.; Fu, K.P. In vitro activity of chloramphenicol and thiamphenicol analogs. Antimicrob. Agents Chemother. 1980, 18, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Syriopoulou, V.P.; Harding, A.L.; Goldman, D.A.; Smith, A.L. In vitro antibacterial activity of fluorinated analogues of chloramphenicol and thiamphenicol. Antimicrob. Agents Chemother. 1981, 19, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.; Palmer, D.; Pratt, B.C.; Hart, C.A. In vitro activity of florfenicol. Eur. J. Clin. Microbiol. Infect. Dis. 1988, 7, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Xia, C.; Shen, J.; Wu, B.; Shen, Z. Characterization of florfenicol resistance among calf pathogenic Escherichia coli. FEMS Microbiol. Lett. 2004, 236, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Braibant, M.; Chevalier, J.; Chaslus-Dancla, E.; Pages, J.-M.; Cloeckaert, A. Structural and functional study of the phenicol-specific efflux pump floR belonging to the major facilitator superfamily. Antimicrob. Agents Chemother. 2005, 49, 2965–2971. [Google Scholar] [CrossRef] [PubMed]
- Doublet, B.; Schwarz, S.; Kehrenberg, C.; Cloeckaert, A. Florfenicol resistance gene floR is part of a novel transposon. Antimicrob. Agents Chemother. 2005, 49, 2106–2108. [Google Scholar] [CrossRef] [PubMed]
- Kehrenberg, C.; Schwarz, S. FexA, a novel Staphylococcus lentus gene encoding resistance to florphenicol and chloramphenicol. Antimicrob. Agents Chemother. 2004, 48, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Kehrenberg, C.; Schwarz, S.; Jacobsen, L.; Hansen, L.H.; Vester, B. A new mechanism for chloramphenicol, florphenicol and clindamycin resistance: Methylation of 23S ribosomal RNA at A2503. Mol. Microbiol. 2005, 57, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.A.; Deprez, P.; H’Haese, E.; Nelis, H.J.; Van den Bossche, W.; De Leenheer, P. Pharmacokinetics of florphenicol in cerebrospinal fluid and plasma of calves. Antimicrob. Agents Chemother. 1997, 41, 1991–1995. [Google Scholar] [PubMed]
- Freeman, K.B. Action of the N-trifluoroacetyl analogue of D-chloramphenicol. Antimicrob. Agents Chemother. 1977, 11, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Butler, P.D.; Sims, P.F.; Wild, D.G. Effects of iodoaphenicol on ribosome assembly in two strains of Escherichia coli. J. Gen. Microbiol. 1982, 128, 997–1001. [Google Scholar] [PubMed]
- Sonenberg, N.; Wilchekt, M.; Zamir, A. Mapping of Escherichia coli ribosomal components involved in peptidyl transferase activity. Proc. Nat. Acad. Sci. USA 1973, 70, 1423–1426. [Google Scholar] [CrossRef] [PubMed]
- Hazra, B.G.; Pore, V.S.; Dey, S.K.; Data, S.; Darokar, M.P.; Saikia, D.; Khanuga, S.P.S.; Thakur, A.P. Bile acid amides derived from chiral amino alcohols: Novel antimicrobials and antifungals. Bioorg. Med. Chem. Lett. 2004, 14, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Coutsogeorgopoulos, C. On the mechanism of action of chloramphenicol in protein synthesis. Biochim. Biophys. Acta 1966, 129, 214–217. [Google Scholar] [CrossRef]
- Vince, R.; Almquist, R.C.; Ritter, C.L.; Daluge, S. Chloramphenicol binding site with analogues of chloramphenicol and puromycin. Antimicrob. Agents Chemother. 1975, 8, 439–443. [Google Scholar] [CrossRef] [PubMed]
- McFarlan, S.C.; Vince, R. Inhibition of peptidyltransferase and possible mode of action of a dipeptidyl chloramphenicol analogue. Biochem. Biophys. Res. Commun. 1984, 122, 748–754. [Google Scholar] [CrossRef]
- Drainas, D.; Mamos, P.; Coutsogeorgopoulos, C. Aminoacyl analogs of chloramphenicol: Examination of the kinetics of inhibition of peptide bond formation. J. Med. Chem. 1993, 36, 3542–3545. [Google Scholar] [CrossRef] [PubMed]
- Michelinaki, M.; Mamos, P.; Coutsogeorgopoulos, C.; Kalpaxis, D.L. Aminoacyl and peptidyl analogs of chloramphenicol as slow-binding inhibitors of ribosomal peptidyltransferase: A new approach for evaluating their potency. Mol. Pharmacol. 1997, 51, 139–146. [Google Scholar] [PubMed]
- Johansson, D.; Jessen, C.H.; Pøhlsgaard, J.; Jensen, K.B.; Vester, B.; Pederden, E.B.; Nielsen, P. Design, synthesis and ribosome binding of chloramphenicol nucleotide and intercalator conjugates. Bioorg. Med. Chem. Lett. 2005, 15, 2079–2083. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, O.N.; Kourelis, T.G.; Mamos, P.; Magoulas, G.E.; Kalpaxis, D.L. Insights into the chloramphenicol inhibition effect on peptidyl transferase activity, using two new analogs of the drug. Open Enz. Inhib. J. 2011, 4, 1–10. [Google Scholar] [CrossRef]
- Mamos, P.; Krokidis, M.G.; Papadas, A.; Karahalios, P.; Starosta, A.L.; Wilson, D.N.; Kalpaxis, D.L.; Dinos, P. On the use of antibiotic chloramphenicol to target polypeptide chain mimics to the ribosomal exit tunnel. Biochimie 2013, 95, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N. Peptides in the ribosomal tunnel talk back. Mol. Cell 2011, 41, 247–248. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.G.; Roy, R.N.; Lomakin, I.B.; Florin, T.; Mankin, A.S.; Steitz, T.A. Structures of proline-rich peptides bound to the ribosome reveal a common mechanism of protein synthesis inhibition. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. The role of outer membrane and efflux pumps on the resistance of Gram-negative bacteria. Can we improve drug access? Drug Resist. Updat. 1998, 1, 93–98. [Google Scholar] [CrossRef]
- Igarashi, K.; Kashiwagi, K. Characteristics of cellular polyamine transport in prokaryotes and eukaruotes. Plant Physiol. Biochem. 2010, 48, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Poolin, R.; Casero, R.A.; Soulet, D. Recent advances in the molecular biology of metazoan polyamine transport. Amino Acids 2012, 42, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Žemlička, J.; Bhuta, A. Sparsophenicol: A new synthetic hybrid antibiotic inhibiting peptide synthesis. J. Med. Chem. 1982, 25, 1123–1125. [Google Scholar] [CrossRef] [PubMed]
- Žemlička, J.; Fernandez-Moyano, M.C.; Ariatti, M.; Zurenko, G.E.; Grady, J.E.; Ballesta, J.P.G. Hybrids of antibiotics inhibiting protein synthesis: Synthesis and biological activity. J. Med. Chem. 1993, 36, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Κim, H.-J.; Lee, J.; Yu, J. Enhanced binding affinity of neomycin-chloramphenicol (or linezolid) conjugates to A-site model of 16S ribosomal RNA. Bull. Korean Chem. Soc. 2006, 27, 1664–1666. [Google Scholar]
- Berkov-Zrihen, Y.; Green, K.D.; Labby, L.J.; Freldman, M.; Garneau-Tsodikova, S.; Fridman, M. Synthesis and evaluation of hetero- and homodimers of ribosome targeting antibiotics: Antimicrobial activity, in vitro inhibition of translation, and drug resistance. J. Med. Chem. 2013, 21, 3624–3631. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, O.N.; Papadopoulos, G.; Kouvela, E.C.; Kalpaxis, D.L. Clindamycin binding to ribosomes revisited: Foοtprinting and computational detection of two binding sites within the peptidyl transferase center. Pharmazie 2013, 36, 1239–1244. [Google Scholar]













© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dinos, G.P.; Athanassopoulos, C.M.; Missiri, D.A.; Giannopoulou, P.C.; Vlachogiannis, I.A.; Papadopoulos, G.E.; Papaioannou, D.; Kalpaxis, D.L. Chloramphenicol Derivatives as Antibacterial and Anticancer Agents: Historic Problems and Current Solutions. Antibiotics 2016, 5, 20. https://doi.org/10.3390/antibiotics5020020
Dinos GP, Athanassopoulos CM, Missiri DA, Giannopoulou PC, Vlachogiannis IA, Papadopoulos GE, Papaioannou D, Kalpaxis DL. Chloramphenicol Derivatives as Antibacterial and Anticancer Agents: Historic Problems and Current Solutions. Antibiotics. 2016; 5(2):20. https://doi.org/10.3390/antibiotics5020020
Chicago/Turabian StyleDinos, George P., Constantinos M. Athanassopoulos, Dionissia A. Missiri, Panagiota C. Giannopoulou, Ioannis A. Vlachogiannis, Georgios E. Papadopoulos, Dionissios Papaioannou, and Dimitrios L. Kalpaxis. 2016. "Chloramphenicol Derivatives as Antibacterial and Anticancer Agents: Historic Problems and Current Solutions" Antibiotics 5, no. 2: 20. https://doi.org/10.3390/antibiotics5020020