

Laboratory Evolution of Antimicrobial Resistance in Bacteria to Develop Rational Treatment Strategies

Abstract
:1. Introduction
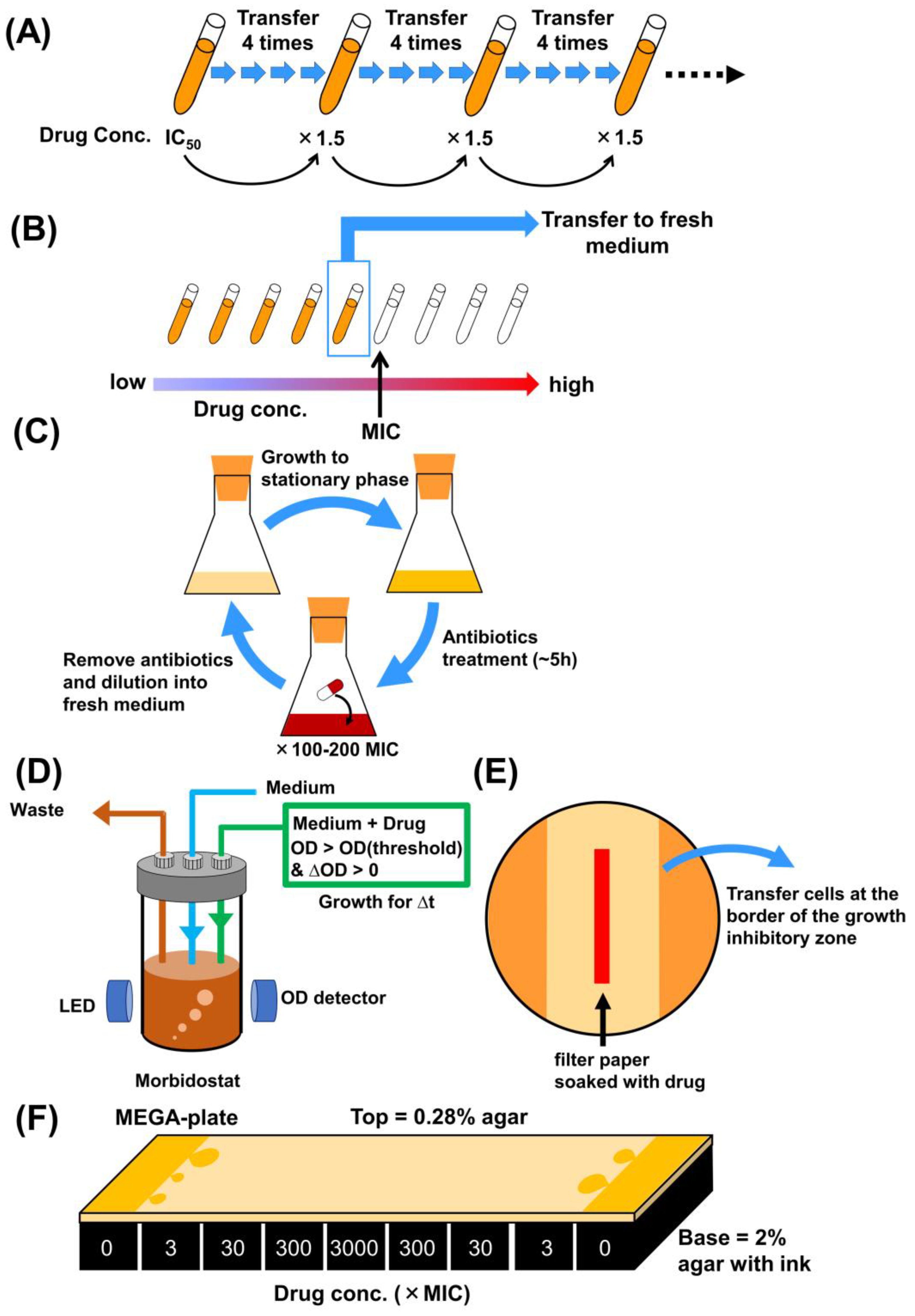
2. Methodologies of Laboratory Evolution
3. Insights into AMR Mechanisms from Laboratory Evolution
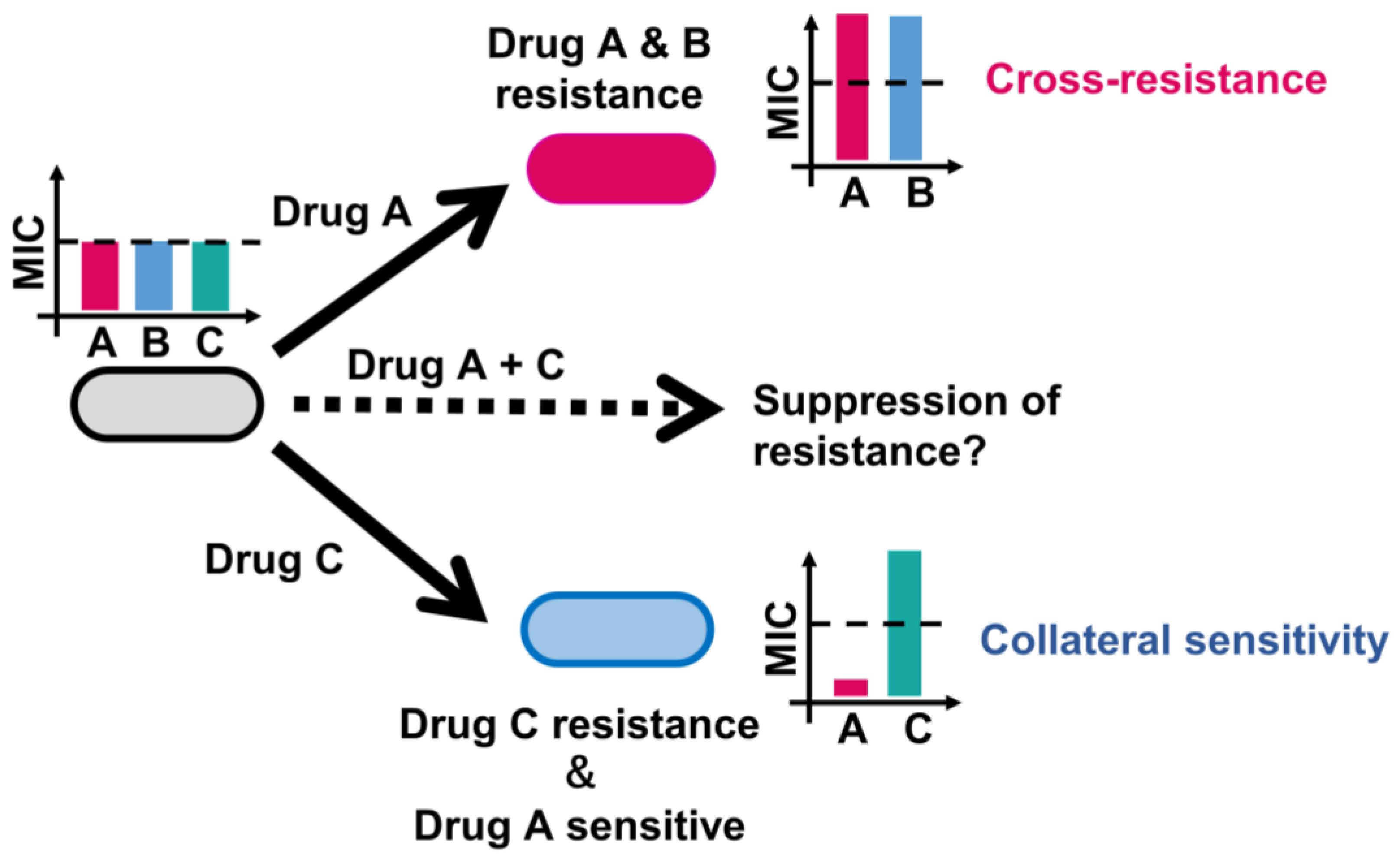
3.1. Genotype-Phenotype Relationships in AMR Evolution
3.2. Identified Key Genes Conferring Cross-Resistance and Collateral Sensitivity in E. coli
3.3. Costs of AMR Evolution
3.4. Impact of Multidrug Combinations on AMR Evolution
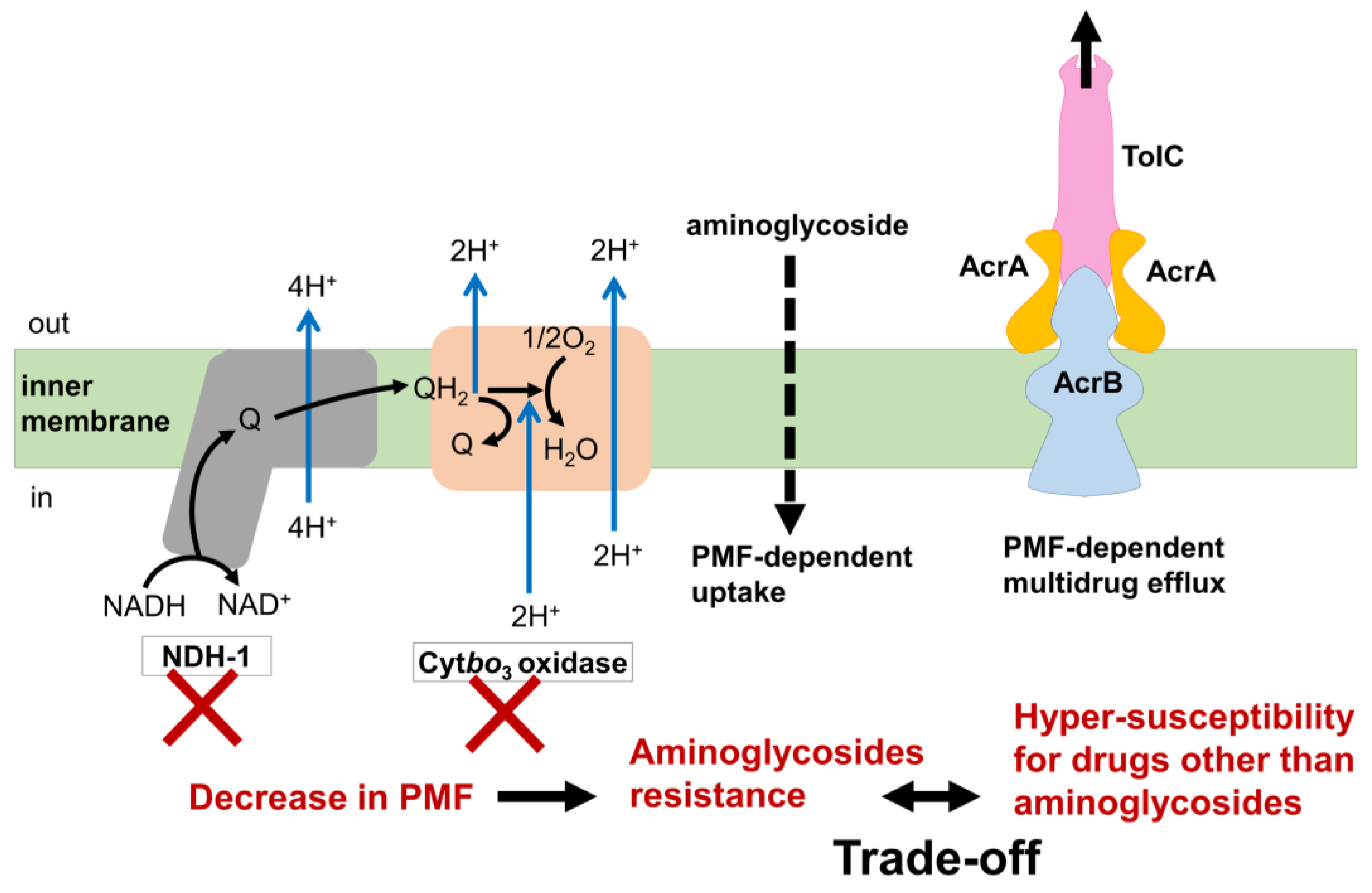
3.5. Mechanism of the Trade-Off between AMR Evolution
3.6. Population Dynamics of AMR Evolution
3.7. AMR Evolution in Spatially Structured Environments
3.8. Fitness Landscape and AMR Evolution
3.9. Antibiotic Tolerance and Persistence Development
4. Designing Rational Treatment Strategies through Laboratory Evolution
4.1. Collateral Sensitivity as a Potential Strategy for Designing Rational Antibiotic Treatment
4.2. Collateral Sensitivity Cycling
4.3. Sequential Drug Regimens Based on Collateral Sensitivities
4.4. Optimization of Antibiotic Treatment for Chronic Infections by Targeting Phenotypic States
4.5. Suppression of Tolerance Acquisition by Cycling Antibiotics with Different Metabolic Dependencies
4.6. Long-Term Clearance Efficacy of Drug Combinations
4.7. Clinical Evidence Supporting the Efficacy of Antibiotic Combination Therapy Involving Aminoglycosides Is Substantial
4.8. Application of Antimicrobial Peptides (AMPs) for Combating AMR

{kind=link}
{kind=link}
{kind=link}
| Strategy | Advantages | Disadvantages | Clinical Trial |
|---|---|---|---|
| Combination of drugs with collateral sensitivity [16,65,82] | Reduce the supply of effective mutations. The trade-off relationship between the drug pair corners pathogens into an evolutionary dead end. | The overall benefits of combinations are not always evident in routine clinical outcomes or from single trials, necessitating a more comprehensive synthesis of clinical data. See Section 4.7. | Positive outcomes have been reported against, e.g., E. coloacae [112], N. gonorrhoear [113], and multidrug-resistant Gram-negative bacteria [110]. Discrepant results were also reported against various pathogens, including S. aureus, Enterobacteriaceae, and P. aeruginosa [114] |
| Collateral sensitivity cycling [15,98] | This strategy upgrades the previous drug-cycling strategy dependent on unreliable fitness costs. Instead, this strategy relies on limiting the evolution of drug resistance. | The potential therapeutic advantage might be overemphasized, with genetic divergence identified as the underlying factor influencing diverse responses, leading to either heightened or diminished resistance to subsequent drugs [26]. | A European cluster-randomized crossover study determined that the practice of antibiotic cycling does not lead to a reduction in the prevalence of antibiotic-resistant Gram-negative bacteria carriage among patients admitted to intensive care units [99]. |
| Cycling antibiotics with different metabolic dependencies [107] | This strategy can interrupt the evolution of tolerance. | Clinical studies are essential to validate its efficacy in real-world hospital settings. | To date, no relevant clinical trial has been reported yet. |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Wellcome Trust and HM Government: London, UK, 2016.
- Roope, L.S.J.; Smith, R.D.; Pouwels, K.B.; Buchanan, J.; Abel, L.; Eibich, P.; Butler, C.C.; Tan, P.S.; Sarah Walker, A.; Robotham, J.V.; et al. The Challenge of Antimicrobial Resistance: What Economics Can Contribute. Science 2019, 364, eaau4679. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Schrader, S.M.; Vaubourgeix, J.; Nathan, C. Biology of Antimicrobial Resistance and Approaches to Combat It. Sci. Transl. Med. 2020, 12, aaz6992. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q.; Helaine, S.; Lewis, K.; Ackermann, M.; Aldridge, B.; Andersson, D.I.; Brynildsen, M.P.; Bumann, D.; Camilli, A.; Collins, J.J.; et al. Definitions and Guidelines for Research on Antibiotic Persistence. Nat. Rev. Microbiol. 2019, 17, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Nicoloff, H.; Hjort, K. Mechanisms and Clinical Relevance of Bacterial Heteroresistance. Nat. Rev. Microbiol. 2019, 17, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Band, V.I.; Hufnagel, D.A.; Jaggavarapu, S.; Sherman, E.X.; Wozniak, J.E.; Satola, S.W.; Farley, M.M.; Jacob, J.T.; Burd, E.M.; Weiss, D.S. Antibiotic Combinations That Exploit Heteroresistance to Multiple Drugs Effectively Control Infection. Nat. Microbiol. 2019, 4, 1627–1635. [Google Scholar] [CrossRef]
- LaFleur, M.D.; Qi, Q.; Lewis, K. Patients with Long-Term Oral Carriage Harbor High-Persister Mutants of Candida Albicans. Antimicrob. Agents Chemother. 2010, 54, 39–44. [Google Scholar] [CrossRef]
- Vulin, C.; Leimer, N.; Huemer, M.; Ackermann, M.; Zinkernagel, A.S. Prolonged Bacterial Lag Time Results in Small Colony Variants That Represent a Sub-Population of Persisters. Nat. Commun. 2018, 9, 4074. [Google Scholar] [CrossRef]
- Woodford, N.; Ellington, M.J. The Emergence of Antibiotic Resistance by Mutation. Clin. Microbiol. Infect. 2007, 13, 5–18. [Google Scholar] [CrossRef]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular Mechanisms of Antibiotic Resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Furusawa, C.; Horinouchi, T.; Maeda, T. Toward Prediction and Control of Antibiotic-Resistance Evolution. Curr. Opin. Biotechnol. 2018, 54, 45–49. [Google Scholar] [CrossRef]
- Yelin, I.; Kishony, R. Antibiotic Resistance. Cell 2018, 172, 1136–1136.e1. [Google Scholar] [CrossRef] [PubMed]
- Lopatkin, A.J.; Bening, S.C.; Manson, A.L.; Stokes, J.M.; Kohanski, M.A.; Badran, A.H.; Earl, A.M.; Cheney, N.J.; Yang, J.H.; Collins, J.J. Clinically Relevant Mutations in Core Metabolic Genes Confer Antibiotic Resistance. Science 2021, 371, eaba0862. [Google Scholar] [CrossRef] [PubMed]
- Imamovic, L.; Sommer, M.O.A. Use of Collateral Sensitivity Networks to Design Drug Cycling Protocols That Avoid Resistance Development. Sci. Transl. Med. 2013, 5, 204ra132. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lieberman, T.D.; Kishony, R. Alternating Antibiotic Treatments Constrain Evolutionary Paths to Multidrug Resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 14494–14499. [Google Scholar] [CrossRef] [PubMed]
- Nichol, D.; Jeavons, P.; Fletcher, A.G.; Bonomo, R.A.; Maini, P.K.; Paul, J.L.; Gatenby, R.A.; Anderson, A.R.A.; Scott, J.G. Steering Evolution with Sequential Therapy to Prevent the Emergence of Bacterial Antibiotic Resistance. PLoS Comput. Biol. 2015, 11, e1004493. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Hernandez, A.; Plucain, J.; Gori, F.; Pena-Miller, R.; Reding, C.; Jansen, G.; Schulenburg, H.; Gudelj, I.; Beardmore, R. Using a Sequential Regimen to Eliminate Bacteria at Sublethal Antibiotic Dosages. PLoS Biol. 2015, 13, e1002104. [Google Scholar] [CrossRef] [PubMed]
- Pál, C.; Papp, B.; Lázár, V. Collateral Sensitivity of Antibiotic-Resistant Microbes. Trends Microbiol. 2015, 23, 401–407. [Google Scholar] [CrossRef]
- Lázár, V.; Pal Singh, G.; Spohn, R.; Nagy, I.; Horváth, B.; Hrtyan, M.; Busa-Fekete, R.; Bogos, B.; Méhi, O.; Csörgo, B.; et al. Bacterial Evolution of Antibiotic Hypersensitivity. Mol. Syst. Biol. 2013, 9, 700. [Google Scholar] [CrossRef]
- Lázár, V.; Nagy, I.; Spohn, R.; Csörgő, B.; Györkei, Á.; Nyerges, Á.; Horváth, B.; Vörös, A.; Busa-Fekete, R.; Hrtyan, M.; et al. Genome-Wide Analysis Captures the Determinants of the Antibiotic Cross-Resistance Interaction Network. Nat. Commun. 2014, 5, 4352. [Google Scholar] [CrossRef]
- Suzuki, S.; Horinouchi, T.; Furusawa, C. Prediction of Antibiotic Resistance by Gene Expression Profiles. Nat. Commun. 2014, 5, 5792. [Google Scholar] [CrossRef]
- Baym, M.; Lieberman, T.D.; Kelsic, E.D.; Chait, R.; Gross, R.; Yelin, I.; Kishony, R. Spatiotemporal Microbial Evolution on Antibiotic Landscapes. Science 2016, 353, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.; Trebosc, V.; Kemmer, C.; Rosenstiel, P.; Beardmore, R.; Schulenburg, H.; Jansen, G. Alternative Evolutionary Paths to Bacterial Antibiotic Resistance Cause Distinct Collateral Effects. Mol. Biol. Evol. 2017, 34, 2229–2244. [Google Scholar] [CrossRef]
- Imamovic, L.; Ellabaan, M.M.H.; Dantas Machado, A.M.; Citterio, L.; Wulff, T.; Molin, S.; Krogh Johansen, H.; Sommer, M.O.A. Drug-Driven Phenotypic Convergence Supports Rational Treatment Strategies of Chronic Infections. Cell 2018, 172, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Nichol, D.; Rutter, J.; Bryant, C.; Hujer, A.M.; Lek, S.; Adams, M.D.; Jeavons, P.; Anderson, A.R.A.; Bonomo, R.A.; Scott, J.G. Antibiotic Collateral Sensitivity Is Contingent on the Repeatability of Evolution. Nat. Commun. 2019, 10, 334. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Iwasawa, J.; Kotani, H.; Sakata, N.; Kawada, M.; Horinouchi, T.; Sakai, A.; Tanabe, K.; Furusawa, C. High-Throughput Laboratory Evolution Reveals Evolutionary Constraints in Escherichia coli. Nat. Commun. 2020, 11, 5970. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The Roles of Mutation, Inbreeding, Crossbreeding, and Selection in Evolution. Proc. Sixth Int. Congr. Genet. 1932, 1, 356–366. [Google Scholar]
- Kauffman, S.; Levin, S. Towards a General Theory of Adaptive Walks on Rugged Landscapes Section of Ecology and Systematics, and Ecosystems Research Center. J. Theor. Biol 1987, 128, 11–45. [Google Scholar] [CrossRef]
- Furusawa, C.; Kaneko, K. Formation of Dominant Mode by Evolution in Biological Systems. Phys. Rev. E 2018, 97, 042410. [Google Scholar] [CrossRef]
- Sato, T.U.; Kaneko, K. Evolutionary Dimension Reduction in Phenotypic Space. Phys. Rev. Res. 2020, 2, 013197. [Google Scholar] [CrossRef]
- Marguerat, S.; Schmidt, A.; Codlin, S.; Chen, W.; Aebersold, R.; Bähler, J. Quantitative Analysis of Fission Yeast Transcriptomes and Proteomes in Proliferating and Quiescent Cells. Cell 2012, 151, 671–683. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Murakami, Y.; Tsuru, S.; Ying, B.W.; Yomo, T. Growth Rate-Coordinated Transcriptome Reorganization in Bacteria. BMC Genom. 2013, 14, 808. [Google Scholar] [CrossRef]
- Schmidt, A.; Kochanowski, K.; Vedelaar, S.; Ahrné, E.; Volkmer, B.; Callipo, L.; Knoops, K.; Bauer, M.; Aebersold, R.; Heinemann, M. The Quantitative and Condition-Dependent Escherichia coli Proteome. Nat. Biotechnol. 2016, 34, 104–110. [Google Scholar] [CrossRef]
- Charlebois, D.A. Quantitative Systems-Based Prediction of Antimicrobial Resistance Evolution. NPJ Syst. Biol. Appl. 2023, 9, 40. [Google Scholar] [CrossRef]
- Oz, T.; Guvenek, A.; Yildiz, S.; Karaboga, E.; Tamer, Y.T.; Mumcuyan, N.; Ozan, V.B.; Senturk, G.H.; Cokol, M.; Yeh, P.; et al. Strength of Selection Pressure Is an Important Parameter Contributing to the Complexity of Antibiotic Resistance Evolution. Mol. Biol. Evol. 2014, 31, 2387–2401. [Google Scholar] [CrossRef] [PubMed]
- Horinouchi, T.; Minamoto, T.; Suzuki, S.; Shimizu, H.; Furusawa, C. Development of an Automated Culture System for Laboratory Evolution. J. Lab. Autom. 2014, 19, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, H.A.; Gallie, J.; Taylor, S.; Kerr, B. Evolutionary Rescue from Extinction Is Contingent on a Lower Rate of Environmental Change. Nature 2013, 494, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Toprak, E.; Veres, A.; Yildiz, S.; Pedraza, J.M.; Chait, R.; Paulsson, J.; Kishony, R. Building a Morbidostat: An Automated Continuous-Culture Device for Studying Bacterial Drug Resistance under Dynamically Sustained Drug Inhibition. Nat. Protoc. 2013, 8, 555–567. [Google Scholar] [CrossRef]
- Maeda, T.; Kawada, M.; Sakata, N.; Kotani, H.; Furusawa, C. Laboratory Evolution of Mycobacterium on Agar Plates for Analysis of Resistance Acquisition and Drug Sensitivity Profiles. Sci. Rep. 2021, 11, 15136. [Google Scholar] [CrossRef]
- Toprak, E.; Veres, A.; Michel, J.-B.; Chait, R.; Hartl, D.L.; Kishony, R. Evolutionary Paths to Antibiotic Resistance under Dynamically Sustained Drug Selection. Nat. Genet. 2011, 44, 101–105. [Google Scholar] [CrossRef]
- Liu, P.C.; Lee, Y.T.; Wang, C.Y.; Yang, Y.T. Design and Use of a Low Cost, Automated Morbidostat for Adaptive Evolution of Bacteria under Antibiotic Drug Selection. J. Vis. Exp. 2016, 2016, e54426. [Google Scholar] [CrossRef]
- Fridman, O.; Goldberg, A.; Ronin, I.; Shoresh, N.; Balaban, N.Q. Optimization of Lag Time Underlies Antibiotic Tolerance in Evolved Bacterial Populations. Nature 2014, 513, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bergh, B.; Michiels, J.E.; Wenseleers, T.; Windels, E.M.; Vanden Boer, P.; Kestemont, D.; Meester, L.d.; Verstrepen, K.J.; Verstraeten, N.; Fauvart, M.; et al. Frequency of Antibiotic Application Drives Rapid Evolutionary Adaptation of Escherichia coli Persistence. Nat. Microbiol. 2016, 1, 16020. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, B.; Schramke, H.; Michiels, J.E.; Kimkes, T.E.P.; Radzikowski, J.L.; Schimpf, J.; Vedelaar, S.R.; Burschel, S.; Dewachter, L.; Lončar, N.; et al. Mutations in Respiratory Complex I Promote Antibiotic Persistence through Alterations in Intracellular Acidity and Protein Synthesis. Nat. Commun. 2022, 13, 1–18. [Google Scholar] [CrossRef]
- Mechler, L.; Herbig, A.; Paprotka, K.; Fraunholz, M.; Nieselt, K.; Bertram, R. A Novel Point Mutation Promotes Growth Phase-Dependent Daptomycin Tolerance in Staphylococcus Aureus. Antimicrob. Agents Chemother. 2015, 59, 5366–5376. [Google Scholar] [CrossRef] [PubMed]
- Michiels, J.E.; Bergh, B.V.D.; Verstraeten, N.; Fauvart, M.; Michiels, J. In Vitro Emergence of High Persistence upon Periodic Aminoglycoside Challenge in the ESKAPE Pathogens. Antimicrob. Agents Chemother. 2016, 60, 4630–4637. [Google Scholar] [CrossRef] [PubMed]
- Nicoloff, H.; Hjort, K.; Levin, B.R.; Andersson, D.I. The High Prevalence of Antibiotic Heteroresistance in Pathogenic Bacteria Is Mainly Caused by Gene Amplification. Nat. Microbiol. 2019, 4, 504–514. [Google Scholar] [CrossRef]
- Wang, Y.; Bojer, M.S.; George, S.E.; Wang, Z.; Jensen, P.R.; Wolz, C.; Ingmer, H. Inactivation of TCA Cycle Enhances Staphylococcus Aureus Persister Cell Formation in Stationary Phase. Sci. Rep. 2018, 8, 10849. [Google Scholar] [CrossRef]
- Cheng, Z.-X.; Yang, M.J.; Peng, B.; Peng, X.; Lin, X.; Li, H. The Depressed Central Carbon and Energy Metabolisms Is Associated to the Acquisition of Levofloxacin Resistance in Vibrio Alginolyticus. J. Proteomics 2018, 181, 83–91. [Google Scholar] [CrossRef]
- Su, Y.-B.; Peng, B.; Li, H.; Cheng, Z.; Zhang, T.; Zhu, J.; Li, D.; Li, M.; Ye, J.; Du, C.; et al. Pyruvate Cycle Increases Aminoglycoside Efficacy and Provides Respiratory Energy in Bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, E1578–E1587. [Google Scholar] [CrossRef]
- Peng, B.; Su, Y.-B.; Li, H.; Han, Y.; Guo, C.; Tian, Y.M.; Peng, X.X. Exogenous Alanine and/or Glucose plus Kanamycin Kills Antibiotic-Resistant Bacteria. Cell Metab. 2015, 21, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Angst, D.C.; Hall, A.R. The Cost of Antibiotic Resistance Depends on Evolutionary History in Escherichia coli. BMC Evol. Biol. 2013, 13, 163. [Google Scholar] [CrossRef] [PubMed]
- Chevereau, G.; Dravecká, M.; Batur, T.; Guvenek, A.; Ayhan, D.H.; Toprak, E.; Bollenbach, T. Quantifying the Determinants of Evolutionary Dynamics Leading to Drug Resistance. PLoS Biol. 2015, 13, e1002299. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, M.; Enke, T.; Chubukov, V.; Ricci, V.; Piddock, L.; Sauer, U. Metabolic Constraints on the Evolution of Antibiotic Resistance. Mol. Syst. Biol. 2017, 13, 917. [Google Scholar] [CrossRef]
- Zampieri, M.; Zimmermann, M.; Claassen, M.; Sauer, U. Nontargeted Metabolomics Reveals the Multilevel Response to Antibiotic Perturbations. Cell Rep. 2017, 19, 1214–1228. [Google Scholar] [CrossRef]
- Apjok, G.; Boross, G.; Nyerges, Á.; Fekete, G.; Lázár, V.; Papp, B.; Pál, C.; Csörgo, B.; Barlow, M. Limited Evolutionary Conservation of the Phenotypic Effects of Antibiotic Resistance Mutations. Mol. Biol. Evol. 2019, 36, 1601–1611. [Google Scholar] [CrossRef]
- Chait, R.; Craney, A.; Kishony, R. Antibiotic Interactions That Select against Resistance. Nature 2007, 446, 668–671. [Google Scholar] [CrossRef]
- Munck, C.; Gumpert, H.K.; Wallin, A.I.N.; Wang, H.H.; Sommer, M.O.A. Prediction of Resistance Development against Drug Combinations by Collateral Responses to Component Drugs. Sci. Transl. Med. 2014, 6, 262ra156. [Google Scholar] [CrossRef]
- Rodriguez de Evgrafov, M.C.; Faza, M.; Asimakopoulos, K.; Sommer, M.O.A. Systematic Investigation of Resistance Evolution to Common Antibiotics Reveals Conserved Collateral Responses across Common Human Pathogens. Antimicrob. Agents Chemother. 2021, 16, e01273-20. [Google Scholar] [CrossRef]
- Taber, H.W.; Mueller, J.P.; Miller, P.F.; Arrow, A.S. Bacterial Uptake of Aminoglycoside Antibiotics. Microbiol. Rev. 1987, 51, 439–457. [Google Scholar] [CrossRef]
- Allison, K.R.; Brynildsen, M.P.; Collins, J.J. Metabolite-Enabled Eradication of Bacterial Persisters by Aminoglycosides. Nature 2011, 473, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, I.T.; Brown, M.H.; Skurray, R.A. Proton-Dependent Multidrug Efflux Systems. Microbiol. Rev. 1996, 60, 575–608. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Yamaguchi, A. Crystal Structure of Bacterial Multidrug Efflux Transporter AcrB. Tanpakushitsu Kakusan Koso. 2003, 48, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Horinouchi, T.; Furusawa, C. Acceleration and Suppression of Resistance Development by Antibiotic Combinations. BMC Genom. 2017, 18, 328. [Google Scholar] [CrossRef]
- Lee, H.H.; Molla, M.N.; Cantor, C.R.; Collins, J.J. Bacterial Charity Work Leads to Population-Wide Resistance. Nature 2010, 467, 82–85. [Google Scholar] [CrossRef]
- Iwasawa, J.; Maeda, T.; Shibai, A.; Kotani, H.; Kawada, M.; Furusawa, C. Analysis of the Evolution of Resistance to Multiple Antibiotics Enables Prediction of the Escherichia coli Phenotype-Based Fitness Landscape. PLoS Biol. 2022, 20, 18–20. [Google Scholar] [CrossRef]
- Michaux, C.; Ronneau, S.; Giorgio, R.T.; Helaine, S. Antibiotic Tolerance and Persistence Have Distinct Fitness Trade-Offs. PLoS Pathog. 2022, 18, e1010963. [Google Scholar] [CrossRef]
- Meylan, S.; Andrews, I.W.; Collins, J.J. Targeting Antibiotic Tolerance, Pathogen by Pathogen. Cell 2018, 172, 1228–1238. [Google Scholar] [CrossRef]
- Windels, E.M.; Michiels, J.E.; van den Bergh, B.; Fauvart, M.; Michiels, J. Antibiotics: Combatting Tolerance to Stop Resistance. MBio 2019, 10, e02095-19. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, K.; Zhang, H.; Jia, Y.; Wang, Z. Combating Antibiotic Tolerance Through Activating Bacterial Metabolism. Front. Microbiol. 2020, 11, 577564. [Google Scholar] [CrossRef]
- Boeck, L. Antibiotic Tolerance: Targeting Bacterial Survival. Curr. Opin. Microbiol. 2023, 74, 102328. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial Persistence as a Phenotypic Switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef] [PubMed]
- Spoering, A.L.; Vulić, M.; Lewis, K. GlpD and PlsB Participate in Persister Cell Formation in Escherichia coli. J. Bacteriol. 2006, 188, 5136–5144. [Google Scholar] [CrossRef]
- Hansen, S.; Lewis, K.; Vulić, M. Role of Global Regulators and Nucleotide Metabolism in Antibiotic Tolerance in Escherichia coli. Antimicrob. Agents Chemother. 2008, 52, 2718–2726. [Google Scholar] [CrossRef] [PubMed]
- Girgis, H.S.; Harris, K.; Tavazoie, S. Large Mutational Target Size for Rapid Emergence of Bacterial Persistence. Proc. Natl. Acad. Sci. USA 2012, 109, 12740–12745. [Google Scholar] [CrossRef] [PubMed]
- Orman, M.A.; Brynildsen, M.P. Dormancy Is Not Necessary or Sufficient for Bacterial Persistence. Antimicrob. Agents Chemother. 2013, 57, 3230–3239. [Google Scholar] [CrossRef]
- Kotte, O.; Volkmer, B.; Radzikowski, J.L.; Heinemann, M. Phenotypic Bistability in Escherichia coli’s Central Carbon Metabolism. Mol. Syst. Biol. 2014, 10, 736. [Google Scholar] [CrossRef]
- Gutierrez, A.; Jain, S.; Bhargava, P.; Hamblin, M.; Lobritz, M.A.; Collins, J.J. Understanding and Sensitizing Density-Dependent Persistence to Quinolone Antibiotics. Mol. Cell 2017, 68, 1147–1154.e3. [Google Scholar] [CrossRef]
- Shan, Y.; Brown Gandt, A.; Rowe, S.E.; Deisinger, J.P.; Conlon, B.P.; Lewis, K.; Formation, A.P.; Shan, Y.; Gandt, A.B.; Rowe, S.E.; et al. Crossm Escherichia coli. MBio 2017, 8, 1–14. [Google Scholar]
- Zalis, E.A.; Nuxoll, A.S.; Manuse, S.; Clair, G.; Radlinski, L.C.; Conlon, B.; Adkins, J.; Lewis, K. Stochastic Variation in Expression of the Tricarboxylic Acid. Am. Soc. Microbiol. 2019, 10, 01930-19. [Google Scholar]
- Melnikov, S.V.; Stevens, D.L.; Fu, X.; Kwok, H.S.; Zhang, J.T.; Shen, Y.; Sabina, J.; Lee, K.; Lee, H.; Söll, D. Exploiting Evolutionary Trade-Offs for Posttreatment Management of Drug-Resistant Populations. Proc. Natl. Acad. Sci. USA 2020, 117, 17924–17931. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Zhang, Y.; Akama, T.; Lau, A.; Zhou, H.; Hernandez, V.; Mao, W.; Alley, M.R.K.; Sanders, V.; Plattner, J.J. Discovery of a New Boron-Containing Antifungal Agent, 5-Fluoro-1,3-Dihydro-1-Hydroxy-2,1-Benzoxaborole (AN2690), for the Potential Treatment of Onychomycosis Stephen. Society 2006, 49, 4447–4450. [Google Scholar]
- Rock, F.L.; Mao, W.; Yaremchuk, A.; Tukalo, M.; Crépin, T.; Zhou, H.; Zhang, Y.K.; Hernandez, V.; Akama, T.; Baker, S.J.; et al. An Antifungal Agent Inhibits an Aminoacyl-TRNA Synthetase by Trapping TRNA in the Editing Site. Science 2007, 316, 1759–1761. [Google Scholar] [CrossRef]
- Jinna, S.; Finch, J. Spotlight on Tavaborole for the Treatment of Onychomycosis. Drug Des. Devel. Ther. 2015, 9, 6185–6190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Ma, S. Recent Development of Leucyl-TRNA Synthetase Inhibitors as Antimicrobial Agents. Medchemcomm 2019, 10, 1329–1341. [Google Scholar] [CrossRef]
- Si, Y.; Basak, S.; Li, Y.; Merino, J.; Iuliano, J.N.; Walker, S.G.; Tonge, P.J. Antibacterial Activity and Mode of Action of a Sulfonamide-Based Class of Oxaborole Leucyl-TRNA-Synthetase Inhibitors. ACS Infect. Dis. 2019, 5, 1231–1238. [Google Scholar] [CrossRef]
- Palencia, A.; Li, X.; Bu, W.; Choi, W.; Ding, C.Z.; Easom, E.E.; Feng, L.; Hernandez, V.; Houston, P.; Liu, L.; et al. Discovery of Novel Oral Protein Synthesis Inhibitors of Mycobacterium Tuberculosis That Target Leucyl-TRNA Synthetase. Antimicrob. Agents Chemother. 2016, 60, 6271–6280. [Google Scholar] [CrossRef]
- Gudzera, O.I.; Golub, A.G.; Bdzhola, V.G.; Volynets, G.P.; Kovalenko, O.P.; Boyarshin, K.S.; Yaremchuk, A.D.; Protopopov, M.V.; Yarmoluk, S.M.; Tukalo, M.A. Identification of Mycobacterium Tuberculosis Leucyl-TRNA Synthetase (LeuRS) Inhibitors among the Derivatives of 5-Phenylamino-2H-[1,2,4]Triazin-3-One. J. Enzyme Inhib. Med. Chem. 2016, 31, 201–207. [Google Scholar] [CrossRef]
- Gudzera, O.I.; Golub, A.G.; Bdzhola, V.G.; Volynets, G.P.; Lukashov, S.S.; Kovalenko, O.P.; Kriklivyi, I.A.; Yaremchuk, A.D.; Starosyla, S.A.; Yarmoluk, S.M.; et al. Discovery of Potent Anti-Tuberculosis Agents Targeting Leucyl-TRNA Synthetase. Bioorganic Med. Chem. 2016, 24, 1023–1031. [Google Scholar] [CrossRef]
- Li, X.; Hernandez, V.; Rock, F.L.; Choi, W.; Mak, Y.S.L.; Mohan, M.; Mao, W.; Zhou, Y.; Easom, E.E.; Plattner, J.J.; et al. Discovery of a Potent and Specific M. Tuberculosis Leucyl-TRNA Synthetase Inhibitor: (S)-3-(Aminomethyl)-4-Chloro-7-(2-Hydroxyethoxy)Benzo[c][1,2]Oxaborol-1(3H)-Ol (GSK656). J. Med. Chem. 2017, 60, 8011–8026. [Google Scholar] [CrossRef]
- Hu, Q.H.; Liu, R.J.; Fang, Z.P.; Zhang, J.; Ding, Y.Y.; Tan, M.; Wang, M.; Pan, W.; Zhou, H.C.; Wang, E.D. Discovery of a Potent Benzoxaborole-Based Anti-Pneumococcal Agent Targeting Leucyl-TRNA Synthetase. Sci. Rep. 2013, 3, srep02475. [Google Scholar] [CrossRef]
- O’Dwyer, K.; Spivak, A.T.; Ingraham, K.; Min, S.; Holmes, D.J.; Jakielaszek, C.; Rittenhouse, S.; Kwan, A.L.; Livi, G.P.; Sathe, G.; et al. Bacterial Resistance to Leucyl-TRNA Synthetase Inhibitor GSK2251052 Develops during Treatment of Complicated Urinary Tract Infections. Antimicrob. Agents Chemother. 2015, 59, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Monteferrante, C.; Rasina, D.; Leitis, G.; Randall, C.P.; Tomlinson, J.H.; Jirgensons, A.; Goessens, W.H.F.; Hays, J.P.; O’Neill, A.J. A Polymorphism in LeuS Confers Reduced Susceptibility to GSK2251052 in a Clinical Isolate of Staphylococcus Aureus. Antimicrob. Agents Chemother. 2016, 60, 3219–3221. [Google Scholar] [CrossRef] [PubMed]
- Raymond, B. Five Rules for Resistance Management in the Antibiotic Apocalypse, a Road Map for Integrated Microbial Management. Evol. Appl. 2019, 12, 1079–1091. [Google Scholar] [CrossRef]
- de Vos, M.; Müller, B.; Borrell, S.; Black, P.A.; Van Helden, P.D.; Warren, R.M.; Gagneux, S.; Victor, T.C. Putative Compensatory Mutations in the Rpoc Gene of Rifampin-Resistant Mycobacterium Tuberculosis Are Associated with Ongoing Transmission. Antimicrob. Agents Chemother. 2013, 57, 827–832. [Google Scholar] [CrossRef]
- McNally, A.; Oren, Y.; Kelly, D.; Pascoe, B.; Dunn, S.; Sreecharan, T.; Vehkala, M.; Välimäki, N.; Prentice, M.B.; Ashour, A.; et al. Combined Analysis of Variation in Core, Accessory and Regulatory Genome Regions Provides a Super-Resolution View into the Evolution of Bacterial Populations. PLoS Genet. 2016, 12, e1006280. [Google Scholar] [CrossRef]
- Yoshida, M.; Reyes, S.G.; Tsuda, S.; Horinouchi, T.; Furusawa, C.; Cronin, L. Time-Programmable Drug Dosing Allows the Manipulation, Suppression and Reversal of Antibiotic Drug Resistance in Vitro. Nat. Commun. 2017, 8, 15589. [Google Scholar] [CrossRef]
- van Duijn, P.J.; Verbrugghe, W.; Jorens, P.G.; Spöhr, F.; Schedler, D.; Deja, M.; Rothbart, A.; Annane, D.; Lawrence, C.; Nguyen Van, J.C.; et al. The Effects of Antibiotic Cycling and Mixing on Antibiotic Resistance in Intensive Care Units: A Cluster-Randomised Crossover Trial. Lancet Infect. Dis. 2018, 18, 401–409. [Google Scholar] [CrossRef]
- Podnecky, N.L.; Fredheim, E.G.A.; Kloos, J.; Sørum, V.; Primicerio, R.; Roberts, A.P.; Rozen, D.E.; Samuelsen, Ø.; Johnsen, P.J. Conserved Collateral Antibiotic Susceptibility Networks in Diverse Clinical Strains of Escherichia coli. Nat. Commun. 2018, 9, 3673. [Google Scholar] [CrossRef]
- Hernando-Amado, S.; López-Causapé, C.; Laborda, P.; Sanz-García, F.; Oliver, A.; Martínez, J.L. Rapid Phenotypic Convergence towards Collateral Sensitivity in Clinical Isolates of Pseudomonas Aeruginosa Presenting Different Genomic Backgrounds. Microbiol. Spectr. 2023, 11, e0227622. [Google Scholar] [CrossRef]
- Poole, K.; Gotoh, N.; Tsujimoto, H.; Zhao, Q.; Wada, A.; Yamasaki, T.; Neshat, S.; Yamagishi, J.I.; Li, X.Z.; Nishino, T. Overexpression of the MexC-MexD-OprJ Efflux Operon in NfxB-Type Multidrug-Resistant Strains of Pseudomonas Aeruginosa. Mol. Microbiol. 1996, 21, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Zheng, E.J.; Stokes, J.M.; Collins, J.J. Eradicating Bacterial Persisters with Combinations of Strongly and Weakly Metabolism-Dependent Antibiotics. Cell Chem. Biol. 2020, 27, 1544–1552.e3. [Google Scholar] [CrossRef] [PubMed]
- Lopatkin, A.J.; Stokes, J.M.; Zheng, E.J.; Yang, J.H.; Takahashi, M.K.; You, L.; Collins, J.J. Bacterial Metabolic State More Accurately Predicts Antibiotic Lethality than Growth Rate. Nat. Microbiol. 2019, 4, 2109–2117. [Google Scholar] [CrossRef]
- Cornforth, D.M.; Dees, J.L.; Ibberson, C.B.; Huse, H.K.; Mathiesen, I.H.; Kirketerp-Møller, K.; Wolcott, R.D.; Rumbaugh, K.P.; Bjarnsholt, T.; Whiteley, M. Pseudomonas Aeruginosa Transcriptome during Human Infection. Proc. Natl. Acad. Sci. USA 2018, 115, E5125–E5134. [Google Scholar] [CrossRef] [PubMed]
- Kordes, A.; Preusse, M.; Willger, S.D.; Braubach, P.; Jonigk, D.; Haverich, A.; Warnecke, G.; Häussler, S. Genetically Diverse Pseudomonas Aeruginosa Populations Display Similar Transcriptomic Profiles in a Cystic Fibrosis Explanted Lung. Nat. Commun. 2019, 10, 3397. [Google Scholar] [CrossRef] [PubMed]
- Zheng, E.J.; Andrews, I.W.; Grote, A.T.; Manson, A.L.; Alcantar, M.A.; Earl, A.M.; Collins, J.J. Modulating the Evolutionary Trajectory of Tolerance Using Antibiotics with Different Metabolic Dependencies. Nat. Commun. 2022, 13, 2525. [Google Scholar] [CrossRef]
- Lázár, V.; Snitser, O.; Barkan, D.; Kishony, R. Antibiotic Combinations Reduce Staphylococcus Aureus Clearance. Nature 2022, 610, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Tamma, P.D.; Cosgrove, S.E.; Maragakis, L.L. Combination Therapy for Treatment of Infections with Gram-Negative Bacteria. Clin. Microbiol. Rev. 2012, 25, 450–470. [Google Scholar] [CrossRef]
- Sick, A.C.; Tschudin-Sutter, S.; Turnbull, A.E.; Weissman, S.J.; Tamma, P.D. Empiric Combination Therapy for Gram-Negative Bacteremia. Pediatrics 2014, 133, e1148–e1155. [Google Scholar] [CrossRef]
- May, A.K. An Argument for the Use of Aminoglycosides in the Empiric Treatment of Ventilator-Associated Pneumonia. Surg. Infect. 2016, 17, 329–333. [Google Scholar] [CrossRef]
- Bodey, G.P.; Elting, L.S.; Rodriguez, S. Bacteremia Caused by Enterobacter: 15 Years of Experience in a Cancer Hospital. Rev. Infect. Dis. 1991, 13, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Kirkcaldy, R.D.; Weinstock, H.S.; Moore, P.C.; Philip, S.S.; Wiesenfeld, H.C.; Papp, J.R.; Kerndt, P.R.; Johnson, S.; Ghanem, K.G.; Hook, E.W. The Efficacy and Safety of Gentamicin plus Azithromycin and Gemifloxacin plus Azithromycin as Treatment of Uncomplicated Gonorrhea. Clin. Infect. Dis. 2014, 59, 1083–1091. [Google Scholar] [CrossRef]
- Paul, M.; Benuri-Silbiger, I.; Soares-Weiser, K.; Leibovici, L. β Lactam Monotherapy versus β Lactam-Aminoglycoside Combination Therapy for Sepsis in Immunocompetent Patients: Systematic Review and Meta-Analysis of Randomised Trials. Br. Med. J. 2004, 328, 668. [Google Scholar] [CrossRef]
- Wang, N.; Luo, J.; Deng, F.; Huang, Y.; Zhou, H. Antibiotic Combination Therapy: A Strategy to Overcome Bacterial Resistance to Aminoglycoside Antibiotics. Front. Pharmacol. 2022, 13, 839808. [Google Scholar] [CrossRef] [PubMed]
- Jahn, L.J.; Simon, D.; Jensen, M.; Bradshaw, C.; Ellabaan, M.M.H.; Sommer, M.O.A. Compatibility of Evolutionary Responses to Constituent Antibiotics Drive Resistance Evolution to Drug Pairs. Mol. Biol. Evol. 2021, 38, 2057–2069. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Huntelman, N.A.; Bullivant, A.; Chacon-Barahona, J.; Valencia, A.; Ida, N.; Zhou, A.; Kalhori, P.; Bello, G.; Xue, C.; Boyd, S.; et al. The Evolution of Resistance to Synergistic Multi-Drug Combinations Is More Complex than Evolving Resistance to Each Individual Drug Component. Evol. Appl. 2023, 16, 1901–1920. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Yan, Z.B.; Meng, Y.M.; Hong, X.Y.; Shao, G.; Ma, J.J.; Cheng, X.R.; Liu, J.; Kang, J.; Fu, C.Y. Antimicrobial Peptides: Mechanism of Action, Activity and Clinical Potential. Mil. Med. Res. 2021, 8, 48. [Google Scholar] [CrossRef]
- Lei, M.; Jayaraman, A.; Van Deventer, J.A.; Lee, K. Engineering Selectively Targeting Antimicrobial Peptides. Annu. Rev. Biomed. Eng. 2021, 23, 339–357. [Google Scholar] [CrossRef]
- Spohn, R.; Daruka, L.; Lázár, V.; Martins, A.; Vidovics, F.; Grézal, G.; Méhi, O.; Kintses, B.; Számel, M.; Jangir, P.K.; et al. Integrated Evolutionary Analysis Reveals Antimicrobial Peptides with Limited Resistance. Nat. Commun. 2019, 10, 4538. [Google Scholar] [CrossRef]
- Maron, B.; Rolff, J.; Friedman, J.; Hayouka, Z. Antimicrobial Peptide Combination Can Hinder Resistance Evolution. Microbiol. Spectr. 2022, 10, e00973-22. [Google Scholar] [CrossRef]
- Shaer Tamar, E.; Kishony, R. Multistep Diversification in Spatiotemporal Bacterial-Phage Coevolution. Nat. Commun. 2022, 13, 7971. [Google Scholar] [CrossRef] [PubMed]
- Loc-Carrillo, C.; Abedon, S.T. Pros and Cons of Phage Therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Elibe Mba, I.; Innocent Nweze, E. Antimicrobial Peptides Therapy: An Emerging Alternative for Treating Drug-Resistant Bacteria. Yale J. Biol. Med. 2022, 95, 445–463. [Google Scholar]



| Mutation | Cross-Resistance | Collateral Sensitivity |
|---|---|---|
| ompF | Chloramphenicol, Rifampicin, Cefmetazole, Aztreonam, Carbenicillin, Norfloxacin, Phleomycin, DL-3-hydroxynorvaline, Mecillinam, Tetracycline, Furaltadone, Erythromycin, Puromycin | D-Cycloserine |
| rssB | Chloramphenicol, Rifampicin, Cefmetazole, Aztreonam, Acriflavine, Carbenicillin, Phleomycin, Tetracycline, Erythromycin, Puromycin | Protamine Sulfate, D-Cycloserine |
| glpT | Carbenicillin, Fosfomycin, Mitomycin C, Phleomycin, Puromycin | Protamine Sulfate, D-Cycloserine, Erythromycin |
| cyaA | Chloramphenicol, Rifampicin, Cefmetazole, Aztreonam, Acriflavine, Carbenicillin, Fosfomycin, Phleomycin, DL-3-hydroxynorvaline, Mecillinam, Tetracycline, Erythromycin, Puromycin | Vancomycin |
| ycbZ | Chloramphenicol, Aztreonam, Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Erythromycin, Puromycin | D-Cycloserine |
| cyoE | Kanamycin, D-Cycloserine, Phleomycin, DL-3-hydroxynorvaline, Puromycin | Rifampicin, Erythromycin |
| cyoA | Kanamycin, Phleomycin | Vancomycin |
| cyoB | Kanamycin, D-Cycloserine, Phleomycin | Sulfisoxazole, Vancomycin |
| nuoG | Aztreonam, Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Puromycin | Chloramphenicol |
| mipA | Acriflavine, Mitomycin C, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Puromycin | Vancomycin |
| ptsP | Aztreonam, Kanamycin, Phleomycin, DL-3-hydroxynorvaline, Puromycin | D-Cycloserine |
| rfe | Rifampicin, Carbenicillin, Mitomycin C, Phleomycin, Mecillinam | D-Cycloserine |
| purR | Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Puromycin | Sulfisoxazole, D-Cycloserine |
| corA | Chloramphenicol, Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Puromycin | Sulfisoxazole |
| oxyR | DL-3-hydroxynorvaline | Norfloxacin |
| apt | Carbenicillin, Puromycin | D-Cycloserine |
| sdaA | Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Puromycin | D-Cycloserine |
| nfsA | Chloramphenicol, Aztreonam, Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Mecillinam, Nitrofurantoin, Furaltadone, Erythromycin, Puromycin | D-Cycloserine |
| ilvL | Chloramphenicol, Acriflavine, Carbenicillin, Puromycin | Sulfisoxazole |
| gshA | Chloramphenicol, Cefmetazole, Aztreonam, Acriflavine, Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Erythromycin, Puromycin | Sulfisoxazole |
| dacA | Chloramphenicol, Acriflavine, Mitomycin C, Phleomycin | Cefmetazole, Erythromycin |
| frlA | Chloramphenicol, Acriflavine | Vancomycin |
| fusA | Chloramphenicol, Rifampicin, Kanamycin, Acriflavine, Carbenicillin, Sulfisoxazole | Protamine Sulfate, D-Cycloserine, Vancomycin |
| glyT | Chloramphenicol, Aztreonam, Acriflavine, Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Erythromycin, Puromycin | Sulfisoxazole |
| gyrA | Chloramphenicol, Cefmetazole, Aztreonam, Carbenicillin, Norfloxacin, Tetracycline, Erythromycin, Puromycin | Acriflavine, Fosfomycin, D-Cycloserine |
| hisS | Chloramphenicol, Tetracycline | D-Cycloserine |
| iscR | Chloramphenicol, Carbenicillin, Mitomycin C, Puromycin | D-Cycloserin |
| livM | Chloramphenicol, Carbenicillin, Tetracycline\ | Sulfisoxazole |
| lon | Chloramphenicol, Cefmetazole, Acriflavine, Carbenicillin, D-Cycloserine, Phleomycin, DL-3-hydroxynorvaline, Mecillinam, Tetracycline, Erythromycin, Puromycin | Mitomycin C |
| rne | Chloramphenicol, Rifampicin, Cefmetazole, Aztreonam, Acriflavine, Carbenicillin, Mitomycin C, Erythromycin | Sulfisoxazole, Vancomycin |
| rob | Chloramphenicol, Rifampicin, Cefmetazole, Aztreonam, Carbenicillin, Norfloxacin, Mitomycin C, Tetracycline, Erythromycin, Puromycin | D-Cycloserine |
| rpoB | Chloramphenicol, Carbenicillin, Mitomycin C, Amitriptyline, Tetracycline, Puromycin | D-Cycloserine |
| serA | Chloramphenicol, Aztreonam, Carbenicillin, Norfloxacin, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Puromycin | Fosfomycin, D-Cycloserine |
| prlF | Chloramphenicol, Aztreonam, Kanamycin, Carbenicillin, D-Cycloserine, Phleomycin, Mecillinam, Puromycin | Rifampicin |
| yjcO | Carbenicillin, Phleomycin, Tetracycline | Sulfisoxazole, D-Cycloserine |
| acrR | Chloramphenicol, Rifampicin, Cefmetazole, Aztreonam, Acriflavine, Carbenicillin, Mitomycin C, Tetracycline, Promethazine, Nitrofurantoin, Furaltadone, Erythromycin, Puromycin | D-Cycloserine |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeda, T.; Furusawa, C. Laboratory Evolution of Antimicrobial Resistance in Bacteria to Develop Rational Treatment Strategies. Antibiotics 2024, 13, 94. https://doi.org/10.3390/antibiotics13010094
Maeda T, Furusawa C. Laboratory Evolution of Antimicrobial Resistance in Bacteria to Develop Rational Treatment Strategies. Antibiotics. 2024; 13(1):94. https://doi.org/10.3390/antibiotics13010094
Chicago/Turabian StyleMaeda, Tomoya, and Chikara Furusawa. 2024. "Laboratory Evolution of Antimicrobial Resistance in Bacteria to Develop Rational Treatment Strategies" Antibiotics 13, no. 1: 94. https://doi.org/10.3390/antibiotics13010094