5.2.5. Structural Characterization for Compounds (3–47)
The 6-Ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone(3): Prepared from 1-(2,5-dihydroxyphenyl)propan-1-one 1a and 2; yellow solid; mp 167.6–167.9 °C; 1H-NMR (CDCl3, 400 MHz) δ 7.11 (d, J = 10.3 Hz, 1H, H-9), 6.81 (d, J = 10.3 Hz, 1H, H-8), 3.76 (s, 3H, 2-NCH3), 3.47 (s, 3H, 4-NCH3), 3.40 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.34 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (CDCl3,100 MHz) δ 185.0 (1C, C-10), 183.9 (1C, C-7), 171.2 (1C, C-6), 159.0 (1C, C-4a), 152.9 (1C, C-1), 151.5 (1C, C-3), 146.6 (1C, C-10a), 138.7 (1C, C-8), 138.7 (1C, C-9), 121.2 (1C, C-6a), 105.4 (1C, C-10b), 32.0 (1C, 6-CH2CH3), 30.6 (1C, 2-NCH3), 29.5 (1C, 4-NCH3), 12.5 (1C, 6-CH2CH3); HRMS m/z 299.09070 (Calculated for C15H13N3O4 [M]+, 299.09061); purified in column chromatography with dichloromethane: ethyl acetate = 9:1; yield: 84%.
The 2,4,6-Trimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (4): Prepared from 1-(2,5-dihydroxyphenyl)ethan-1-one 1b and 2; yellow solid; mp 197.5–198.5 °C (d); 1H-NMR (CDCl3,400 MHz) δ 7.13 (d, J = 10.5 Hz, 1H, 8-H), 6.83 (d, J = 10.5 Hz, 1H, 9-H), 3.75 (s, 3H, 2-NCH3), 3.47 (s, 3H, 4-NCH3), 2.99 (s, 3H, 6-CH3); 13C-NMR (CDCl3,100 MHz) δ 184.2, 183.4, 166.2, 158.3, 152.3, 150.9, 145.8, 138.4, 138.1, 121.1, 105.2, 30.1, 28.9, 26.6; HRMS m/z 285.0828 (Calculated forC14H11N3O4 [M + H]+: 286.0832); purified by column chromatography, dichloromethane: ethyl acetate = 9:1; yield: 86%.
The 2,4-Dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (5): Prepared from 2,5-dihydroxybenzaldehyde 1c and 2; yellow solid; mp 203.5–205.5 °C (d);1H-NMR (400 MHz, CDCl3) δ 9.30 (s, 1H, 6-H), 7.15 (d, J = 10.5 Hz, 1H, 9-H), 6.88 (d, J = 10.5 Hz, 1H, 8-H), 3.79 (s, 3H, 2-NCH3), 3.51 (s, 3H, 4-NCH3); 13C-NMR (100 MHz, CDCl3) δ 182.8, 182.1, 158.2, 154.8, 153.1, 150.8, 142.9, 140.6, 136.3, 122.6, 106.3, 30.5, 29.1; HRMS m/z 272.0671 (Calculated for C13H9N3O4 [M + H]+: 272.06); purified by column chromatography with a mixture of dichloromethane: ethyl acetate = 9:1; yield: 86%.
The 8-(4-Aminobenzenethio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (6).A suspension of 11 (150.0 mg, 0.33 mmol), iron powder (370 mg, 6.63 mmol) in a 1:1:1 mixture of water/methanol/acetic acid (30 mL) was stirred for 1 h at 50–60 °C. The mixture was neutralized with NaHCO3 and then extracted with ethyl acetate (2 × 15 mL). The organic extract was dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The organic crude was purified using 45g of silica gel 60 (230–400 mesh). The resulting solution was concentrated to dryness under reduced pressure; brown solid; mp > 250 °C; 1H-NMR (400 MHz, CDCl3) δ 7.24 (d, J = 8.5 Hz, 2H, 3′-H y 5′-H), 6.73 (d, J = 8.5 Hz, 2H, 2′-H y 6′-H), 6.22 (s, 1H, 9-H), 4.01 (s, 2H, 4′-NH2), 3.74 (s, 3H, 2-NCH3), 3.42 (s, 3H, 4-NCH3), 3.40 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.35 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3): δ 181.5, 181.2, 170.7, 158.5, 158.1, 152.7, 151.1, 148.9, 147.5, 137.0 (2C), 127.7, 120.8, 116.3 (2C), 113.4, 105.4, 31.7, 30.2, 29.0, 12.2. HRMS m/z 423.1125 (Calculated for C21H19N4O4S[M + H]+: 423.1127); purified by column chromatography with a mixture of dichloromethane: ethyl acetate: petroleum ether = 9:1:1; yield: 31%.
The 8-(4-Acetamidobenzenethio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (7): Prepared from 3 and 4-acetamidothiophenol using general procedure B; orange solid; mp 170.2–173.0 °C; 1H-NMR (400 MHz, CDCl3) δ 7.99 (s, 1H, NHCO), 7.69 (d, J = 8.3 Hz, 2H, 3′-H and 5′-H), 7.43 (d, J = 8.4 Hz, 2H, 2′-H and 6′-H), 6.15 (s, 1H, 9-H), 3.75 (s, 3H, 2-NCH3), 3.42 (s, 3H, 4-NCH3), 3.40 (q, J = 7.5 Hz, 2H, 6-CH2CH3), 2.21 (s, 3H, COCH3), 1.36 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3): δ 181.3, 180.8 (2C), 170.8, 168.9, 158.5, 157.2, 152.7, 151.0, 147.2, 140.6, 136.5 (2C), 127.7, 121.1 (2C), 120.6, 105.4, 31.7, 30.2, 29.1, 24.7, 12.1; HRMS m/z 465.1246 (Calculated for C23H21N4O5S [M + H]+: 465.1233); purified in column chromatography with dichloromethane: ethyl acetate = 3:1; yield: 69%.
The 8-(4-hydroxybenzenethio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone(8): Prepared from 3 and 4-mercaptophenol using general procedure B; orange solid; mp 208–210 °C (d); 1H-NMR (400 MHz, CDCl3) δ 7.38 (d, J = 8.7 Hz, 2H, 3′-H and 5′-H), 6.96 (d, J = 8.7 Hz, 2H,2′-H and 6′-H), 6.21 (s, 1H, 4′-OH), 6.15 (s, 1H, 9-H), 3.77 (s, 3H, 2-NCH3), 3.46 (s, 3H, 4-NCH3), 3.43 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.38 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 181.5, 181.0, 171.0, 159.1, 158.2, 157.6, 155.8, 152.7, 151.0, 147.4, 137.4 (2C), 127.7, 120.8, 117.6 (2C), 117.0, 31.8, 30.3, 29.2, 12.2; HRMS m/z 424.0963 (Calculated for C21H18N3O5S [M + H]+: 424.0967); purified in column chromatography with ethyl acetate: petroleum ether = 9:0.8; yield: 72%.
The 4-((6-ethyl-2,4-dimethyl-1,3,7,10-tetraoxo-1,2,3,4,7,10-hexahydropyrimido[4,5-c]isoquinolin-8-yl)thio)benzonitrile (9): Prepared from 3 and 4-mercaptobenzonitrile using general procedure B; orange solid; mp 203–205 °C; 1H-NMR (400 MHz, CDCl3) δ 7.80 (d, J = 8.0 Hz, 2H, 3′-H and 5′-H), 7.69 (d, J = 8.0 Hz, 2H, 2′-H and 6′-H), 6.21 (s, 1H, 9-H), 3.76 (s, 3H, 2-NCH3), 3.44 (s, 3H, 4-NCH3), 3.41 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.37 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 181.1, 180.2, 171.0, 158.3, 158.2, 154.6, 152.9, 151.0, 146.9, 136.2 (2C), 134.0, 133.7 (2C) 128.4, 120.3, 117.6, 114.6, 105.5, 31.8, 30.2, 29.1, 12.1; HRMS m/z 433.0892 (Calculated for C22H17N4O4S [M + H]+: 433.0971); purified in column chromatography with dichloromethane: ethyl acetate = 9:1; yield: 72%.
Synthesis of 8-(4-carboxibenzenethio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (10). Prepared from 3 and 4-mercaptobenzoic acid; A solution of 3 (150 mg, 0.4909 mmol) and CeCl3∙7H2O (5% mmol respect to 3) in a mix of ethanol: dichloromethane = 1:1 (10 mL), was added dropwise slowly a solution of 4-mercaptobenzoic acid (0.5 equiv.) in ethanol: dichloromethane = 1:1 (30 mL). The reaction mixture was stirred at room temperature for 16 h. The progress of the reaction was followed by thin-layer chromatography (TLC). Then, 10 mL of distilled water and NaOH (0.1 M) are added to the solution until pH 10 is reached. The extractions were carried out with ethyl acetate (10 mL × 2), the precipitate solid, filtered under vacuum, and washed with ethanol (15 mL × 3). Finally, the obtained product was recrystallized from ethanol. Orange solid, mp > 250 °C; 1H-NMR (400 MHz DMSO6) δ 13.29 (s, 1H, 4′-COOH), 8.10 (d, 2H, 3′-H and 5′-H), 7.76 (d, 2H, 2′-H and 6′-H), 6.07 (s, 1H, 9-H), 3.59 (s, 3H, 2-NCH3), 3.30 (q, J = 7.3 Hz, 2H, 6-CH2CH3) 3.23 (s, 3H, 4-NCH3), 1.29 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz DMSO6) δ 181.1, 180.4, 169.3, 167.0, 158.3, 155.1, 152.9, 151.2, 146.4, 135.8, 133.1, 133.0, 131.5 (2C), 127.8 (2C), 120.6, 105.8, 31.4, 30.3, 29.0, 12.3; HRMS m/z 452.0912 (Calculated for C22H18N3O6S [M + H]+: 452.0916); yield: 38%.
The 8-(4-nitrobenzenethio)-6-ethyl-2,4-dimethyl-pyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (11): Prepared from 3 and 4-nitrobenzenethiol using general procedure B; yellow solid; mp 188.8–190.1 °C (d); 1H-NMR (400 MHz, CDCl3) δ 8.34 (d, 2H, 3′-H and 5′-H), 7.75 (d, 2H, 2′-H and 6′-H), 6.24 (s, 1H, 9-H), 3.75 (s, 3H, 2-NCH3), 3.42 (s, 3H, 4-NCH3), 3.40 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.36 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 181.0, 181.0, 170.9, 158.3, 154.3, 152.9, 151.0, 149.1, 146.9, 136.4 (2C), 136.1, 128.6, 125.1 (2C), 120.2, 105.4, 31.8, 30.3, 29.1, 12.1; HRMS m/z 453.0871 (Calculated for C21H17N4O6S [M + H]+: 453.0869); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 10:1:4; yield: 96%.
The 8-(4-dimethylaminobenzenethio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (12): Prepared from 3 and 4-(dimethylamino)benzenethiol using general procedure B; burgundy red; mp 134.6–137.0 °C; 1H-NMR (400 MHz, CDCl3) δ 7.31 (d, J = 8.6 Hz, 2H, 2′-H and 6′-H), 6.75 (d,J = 8.6 Hz, 2H, 3′-H and 5′-H), 6.21 (s, 1H, 9-H), 3.74 (s, 3H, 2-NCH3), 3.43 (s, 3H, 4-NCH3), 3.40 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 3.03 (s, 6H, 4′-N(CH3)2), 1.36 (t, J = 7.3 Hz, 3H, 6-CH2CH3);13C-NMR (100 MHz, CDCl3) δ 181.7, 181.5, 170.7, 158.7, 158.6, 152.8, 151.9, 151.3, 147.7, 136.7 (2C), 127.8, 121.0, 113.6 (2C), 110.6, 105.5, 77.2, 40.3, 31.8, 30.3, 29.2, 12.3; HRMS m/z 451.1447 (Calculated for C23H23N4O4S [M + H]+: 451.1440); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 9:0.5:1; yield: 43%.
The 8-(4-(methylthio)benzenethio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (13): Prepared from 3 and 4-(methylthio)benzenethiol using general procedure B; red solid; mp 181.6–184.0 °C; 1H-NMR (400 MHz, CDCl3) δ 7.41 (d, J = 8.1 Hz, 2H, 2′-H and 6′-H), 7.33 (d, J = 8.2 Hz, 2H, 2′-H and 6′-H), 6.19 (s, 1H, 9-H), 3.75 (s, 3H, 2-NCH3), 3.43 (s, 3H, 4-NCH3), 3.41 (q, J = 7.4 Hz, 2H, 6-CH2CH3), 2.53 (s, 3H, SCH3), 1.36 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 181.33, 180.8, 170.7, 158.4, 156.8, 152.7, 151.1, 147.3, 143.1, 135.8 (2C), 127.9, 127.4 (2C), 122.4, 120.6, 105.4, 31.7, 30.2, 29.1, 15.2, 12.1; HRMS m/z 454.0894 (Calculated for C22H20N3O4S2 [M + H]+: 454.0895); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 3:2:0.5; yield: 46%.
The 6-ethyl-8-(4-ethylbenzenethio)-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (14): Prepared from 3 and 4-ethylbenzenethiol using general procedure B; yellow solid; mp 172–174 °C; 1H-NMR (400 MHz, CDCl3) δ 7.43 (d, J = 7.9 Hz, 2H, 3′-H and 5′-H), 7.33 (d, J = 7.9 Hz, 2H, 2′-H and 6′-H), 6.18 (s, 1H, 9-H), 3.75 (s, 3H, 2-NCH3), 3.43 (s, 3H, 4-NCH3), 3.41 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 2.72 (q, J = 7.6 Hz, 2H, 4′-CH2CH3), 1.37 (t, J = 7.3 Hz, 3H, 4′-CH2CH3), 1.28 (t, J =7.6 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 181.4, 180.9, 170.7, 158.4, 157.2, 152.7, 151.1, 147.4, 135.7 (2C), 130.1 (2C), 127.8, 123.7, 120.6, 105.4, 31.7, 30.2, 29.0, 28.7, 15.3, 12.2; HRMS m/z 436.1253 (Calculated for C23H22N3O4S [M + H]+: 436.1331); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 12:0.5:5; yield: 83%.
The 6-ethyl-2,4-dimethyl-8-((4-trifluoromethyl)phenyl)thio)pyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone(15): Prepared from 3 and 4-(trifluoromethyl)benzenethiol using general procedure B; yellow solid; mp 204–206 °C; 1H-NMR (400 MHz, CDCl3) δ 7.77 (d, J = 8.1 Hz, 2H, 3′-H and 5′-H), 6.96 (d, J = 8.1 Hz, 2H, 2′-H and 6′-H), 6.18 (s, 1H, 9-H), 3.76 (s, 3H, 2-NCH3), 3.43 (s, 3H, 4-NCH3), 3.42 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.37 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 181.2, 181.4, 170.9, 158.3, 155.3, 152.8, 151.0, 147.1, 136.1 (2C), 132.8 (q, J = 33.1 Hz, 1C), 132.2, 128.2, 127.2 (q, J = 3.6 Hz, 2C), 123.5 (q, J = 272.8 Hz, 1C), 120.4, 105.5, 31.8, 30.2, 29.1, 12.1; HRMS m/z 476.0814 (Calculated for C22H17F3N3O4S [M + H]+: 476.0812); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 12:0.5:5; yield: 89%.
The 8-(4-(isopropyl)benzenethio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (16): Prepared from 3 and 4-isopropylbenzenethiol using general procedure B; orange solid; mp 134.6–137.0 °C; 1H-NMR (400 MHz, CDCl3) δ 7.43 (d, J = 8.0 Hz, 2H, 3′-H and 5′-H), 7.35 (d, J = 8.0 Hz, 2H, 2′-H and 6′-H), 6.17 (s, 1H, 9-H), 3.74 (s, 3H, 2-NCH3), 3.42 (s, 3H, 4-NCH3), 3.40 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 3.03–2.90 (m, 1H, 4′-CH(CH3)2), 1.36 (t, J = 7.3 Hz, 3H, 6-CH2CH3), 1.28 (d, J = 6.9 Hz, 6H, 4′-CH(CH3)2); 13C-NMR (100 MHz, CDCl3) δ 181.6, 181.0, 170.8, 158.5, 157.3, 152.8, 152.1, 151.2, 147.5, 135.8(2C), 128.7 (2C), 127.9, 123.9, 120.7, 105.5, 34.2, 31.8, 30.2, 29.1, 23.9 (2C), 12.2; HRMS m/z 450.1499 (Calculated for C24H24N3O4S [M + H]+: 450.1488); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 12:0.5:1; yield: 43%.
The 8-(4-(tertbutyl)benzenethio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (17): Prepared from 3 and 4-(tert-butyl)benzenethiol using general procedure B; orange solid, mp 157.6–160.7 °C; 1H-NMR (400 MHz, CDCl3) δ 7.52 (d, J = 8.2 Hz, 2H, 3′-H and 5′-H), 7.45 (d, J = 8.1 Hz, 2H, 2′-H and 6′-H), 6.19 (s, 1H, 9-H), 3.75 (s, 3H, 2-NCH3), 3.43 (s, 3H, 4-NCH3), 3.42 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.37 (t, J = 7.4 Hz, 3H, 6-CH2CH3), 1.36 (s, 9H, 4′-C(CH3)3); 13C-NMR (100 MHz, CDCl3) δ 181.7, 181.0, 170.9, 158.6, 157.3, 154.4, 152.8, 151.2, 147.5, 135.5(2C), 128.0, 127.9, 127.7, 126.2, 123.7, 120.7, 105.5, 35.1, 31.8, 31.4, 31.3, 30.3, 29.2, 12.3; HRMS m/z 464.1653 (Calculated for C25H26N3O4S [M + H]+: 464.1644); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 12:0.5:5; yield: 61%.
The 8-(benzylthio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (18): Prepared from 3 and phenylmethanethiol using general procedure B; orange solid; mp 181.0–182.0 °C; 1H-NMR (400 MHz, CDCl3) δ 7.40–7.28 (m, 5H, C6H5), 6.76 (s, 1H, 9-H), 4.06 (s, 2H, Ph-CH2-S), 3.74 (s, 3H, 2-NCH3), 3.44 (s, 3H, 4-NCH3), 3.36 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.33 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 180.8 (2C), 170.8, 158.5, 154.6, 152.8, 151.2, 147.1, 134.0, 129.1 (2C), 129.0 (2C), 128.2, 127.1, 120.7, 105.4, 35.8, 31.8, 30.2, 29.1, 12.2. HRMS m/z 422.1171 (Calculated for C22H20N3O4S [M + H]+: 422.1175); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 9:1:3; yield: 66%.
The 8-(phenethylthio)-6-ethyl-2,4-dimethyl-pyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (19): Prepared from 3 and 2-phenylethanethiol using general procedure B; yellow solid; mp 170.0–171.0 °C; 1H-NMR (400 MHz, CDCl3) δ 7.33 (t, J = 7.3 Hz, 2H, 3′-H and 5′-H), 7.26–7.24 (m, 3H, 2′-H, 4′-H and 6′-H), 6.70 (s, 1H, 9-H), 3.75 (s, 3H, 2-NCH3), 3.46 (s, 3H, 4-NCH3), 3.37 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 3.13–3.00 (m, 4H, Ph-C2H4S), 1.34 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 180.7 (2C), 170.7, 158.5, 154.9, 152.7, 151.1, 147.0, 138.9, 128.8 (2C), 128.5 (2C), 127.0, 126.5, 120.7, 105.4, 33.7, 32.3, 31.7, 30.2, 29.1, 12.1; HRMS m/z 436.1332 (Calculated for C23H22N3O4S [M + H]+: 436.1331); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 10:1:6; yield: 79%.
The (4-(chlorobenzyl)thio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (20): Prepared from 3 and (4-chlorophenyl)methanethiol using general procedure B; orange solid; mp 191.0–191.8 °C; 1H-NMR (400 MHz, CDCl3) δ 7.33 (m, 4H, 2′-H, 3′-H, 5′-H, and 6′-H), 6.72 (s, 1H, 9-H), 4.02 (s, 2H, Ph-CH2-S), 3.74 (s, 3H, 2-NCH3), 3.45 (s, 3H, 4-NCH3), 3.36 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.33 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 180.7, 180.6, 170.7, 158.4, 154.1, 152.7, 151.0, 146.9, 134.0, 132.4, 130.2 (2C), 129.2 (2C), 127.1, 120.6, 105.4, 35.0, 31.7, 30.2, 29.1, 12.1; HRMS m/z 456.0775 (Calculated for C22H19ClN3O4S [M + H]+: 456.0785); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 10:1:5; yield: 32%.
The 8-(benzo[d]thiazol-2-ylthio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone(21): Prepared from 3 and benzo[d]thiazole-2-thiol using general procedure B; yellow solid; mp > 250 °C; 1H-NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.1 Hz, 1H, 7′-H), 7.89 (d, J = 8.1 Hz, 1H, 4′-H), 7.54 (d, J = 7.6 Hz, 1H, 5′-H), 7.47 (d, J = 7.6 Hz, 1H, 6′-H), 6.25 (s, 1H, 9-H), 3.74 (s, 3H, 2-NCH3), 3.42 (s, 3H, 4-NCH3), 3.38 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.34 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 181.5, 179.9, 170.9, 158.2, 156.7, 153.6, 152.9, 151.0, 150.5, 146.8, 136.9, 131.4, 126.7, 123.6, 121.5, 105.4 31.7, 30.2, 29.1, 12.1; HRMS m/z 465.0690 (Calculated for C22H17N4O4S 2 [M + H]+: 465.0691); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 9:1:3; yield: 72%.
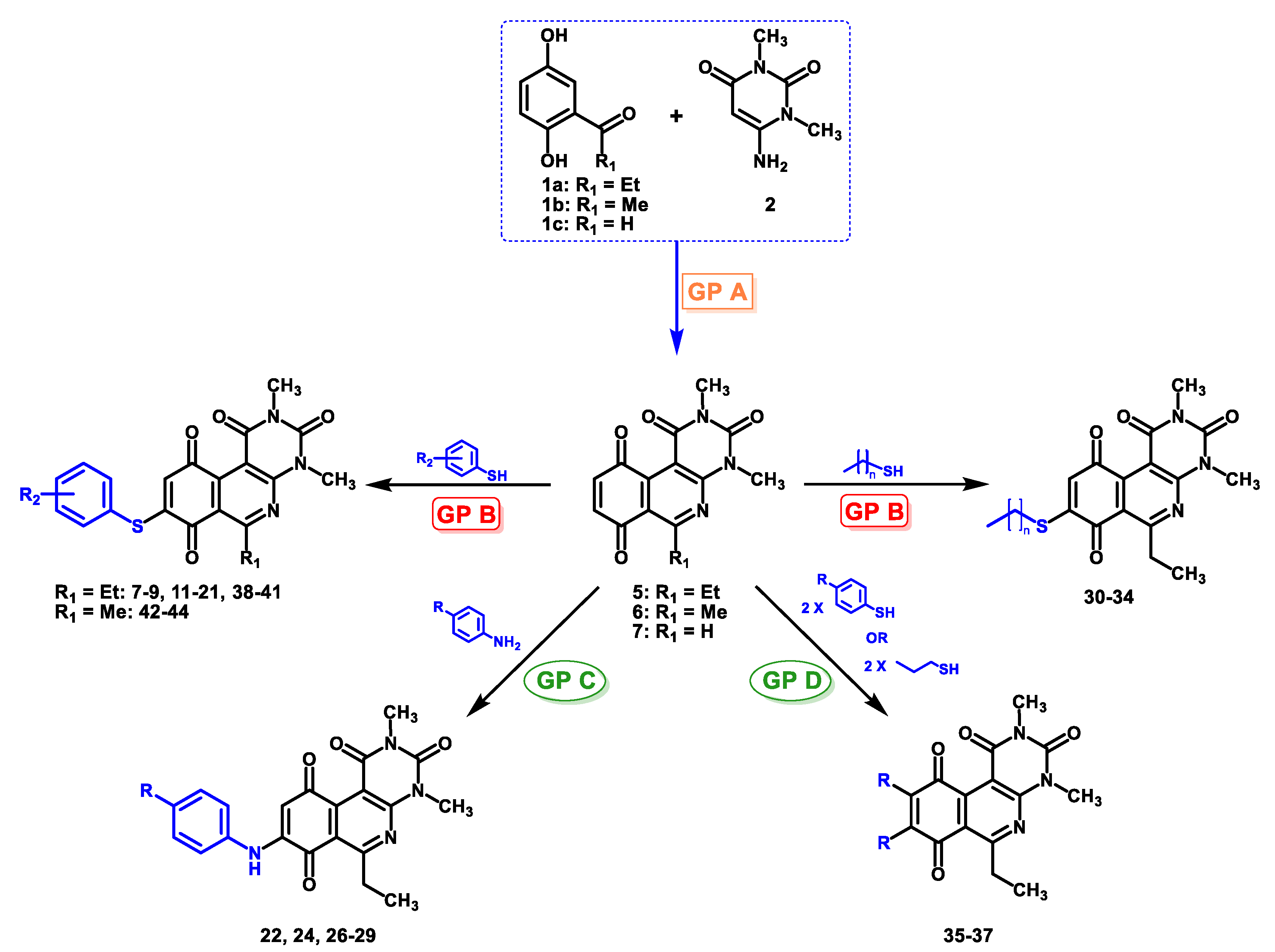
The 8-(phenylamino)-6-ethyl-2,4-dimethyl-pyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (22): Prepared from 3 and aniline using general procedure C; purple solid; mp 189.0–190.0 °C; 1H -NMR (400 MHz, CDCl3) δ 7.60 (s, 1H, NH), 7.42 (t, J = 7.8 Hz, 2H, 3′-H and 5′-H), 7.23 (m, 3H, 2′-H, 4′-H and 6′-H), 6.46 (s, 1H, 9-H), 3.76 (s, 3H, 2-NCH3), 3.47 (s, 3H, 4-NCH3), 3.41 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.37 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 182.2, 180.0, 170.2, 158.7, 153.1, 151.2, 149.3, 144.6, 137.2, 129.8(2C), 125.8, 122.3(2C), 119.5, 105.9, 103.7, 31.8, 30.2, 29.1, 12.1; HRMS m/z 391.1412 (Calculated for C21H19N4O4 [M + H]+: 391.1406); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 1:2:4; yield: 76%.
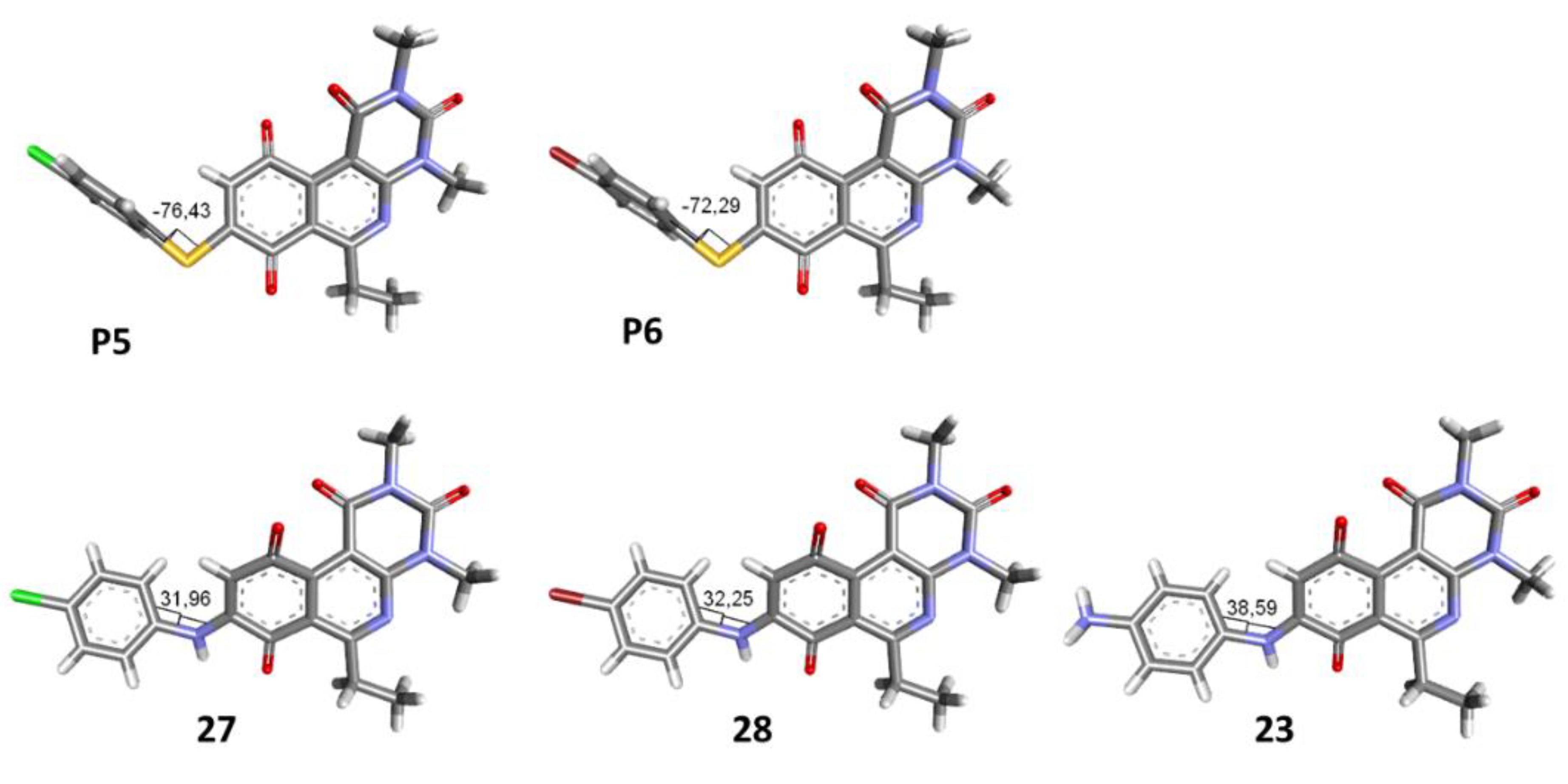
Synthesis of 8-(4-amino-phenylamino)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (23): A solution of 3 (150 mg, 0.4909 mmol) and CeCl3*7H2O (5% mmol respect to 1) in a mix of ethanol: dichloromethane = 1:1 (10 mL), was added dropwise slowly a solution of benzene-1,4-phenylendiamine (26.60 mg, 0.2454 mmol) in ethanol: dichloromethane = 1:1 (30mL). The reaction mixture was stirred at room temperature for 16 h. The progress of the reaction was followed by thin-layer chromatography (TLC). The reaction mixture was concentrated under reduced pressure, and the crude reaction was purified using 30 g of silica gel (70–230 mesh) and a mix of chloroform and ethyl acetate as eluent; green solid; mp > 250 °C; 1H-NMR (400 MHz, CDCl3) δ 7.26 (d, J = 8.3 Hz, 2H, 2′-H and 6′-H), 7.26 (s, 1H, NH), 6.75 (d, J = 8.4 Hz, 2H, 3′-H and 5′-H), 6.23 (s, 1H, 9-H), 3.99 (s, 2H, 4′-NH2), 3.75 (s, 3H, 2-NCH3), 3.44 (s, 3H, 4-NCH3), 3.41 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.36 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 181.5, 181.2, 170.7, 158.5, 158.1, 152.7, 151.1, 148.9, 147.5, 137.0 (2C), 127.7, 120.8, 116.4 (2C), 113.5, 105.4, 31.7, 30.2, 29.0, 12.2; HRMS m/z 406.1528 (Calculated for C21H20N5O4 [M + H]+: 406.1515); purified in column chromatography with chloroform: ethyl acetate = 8:1; yield: 51%.
The 8-(4-(methoxycarbonyl)phenylamino)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (24): Prepared from 3 and methyl 4-aminobenzoate using general procedure C; red solid; mp > 250 °C; 1H-NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8.1 Hz, 2H, 3′-H and 5′-H), 7.76 (s, 1H, NH), 7.31 (d, J = 8.1 Hz, 2H, 2′-H and 6′-H), 6.63 (s, 1H, 9-H), 3.93 (s, 3H, 4′-COOCH3), 3.77 (s, 3H, 2-NCH3), 3.47 (s, 3H, 4-NCH3), 3.41 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.38 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 182.4, 179.6, 170.4, 155.4, 151.9, 143.3, 141.6, 134.5, 132.6 (2C), 131.4, 123.9 (2C), 120.6, 118.9, 110.4, 108.9, 105.5, 57.6, 31.9, 30.2, 29.1, 12.0; HRMS m/z 449.1469 (Calculated for C23H21N4O6 [M + H]+: 449.1461); purified in column chromatography with chloroform: ethyl acetate = 20:1; yield: 42%.
Synthesis of methyl 4-(6-ethyl-2,4-dimethyl-1,3,7,10-tetraoxo-1,2,3,4,7,10-hexahydropyrimido[4,5-c]isoquinolin-8-yl)amino)benzoic acid (25). Prepared from 3 and 4-aminobenzoic acid; A solution of 3 (150 mg, 0.4909 mmol) and CeCl3*7H2O (5% mmol respect to 3) in a mix of ethanol: dichloromethane = 1:1 (10 mL), was added dropwise slowly a solution of 4-aminobenzoic acid (34.40 mg, 0.2506 mmol) in ethanol: dichloromethane = 1:1 (30 mL). The reaction mixture was stirred at room temperature for 16 h. The progress of the reaction was followed by thin-layer chromatography (TLC). The reaction mixture was concentrated under reduced pressure, and the obtained solid was washed three times with 30 mL of dichloromethane. Finally, the solid was purified using 10 g of silica gel (70–230 mesh) and ethyl acetate as eluent; red solid; mp > 250 °C; 1H-NMR (400 MHz DMSO-d6) δ 9.47 (s, 1H, NH), 7.97 (d, J = 8.6 Hz, 2H, 3′-H and 5′-H), 7.54 (d, J = 8.7 Hz, 2H, 2′-H and 6′-H), 6.40 (s, 1H, 9-H), 3.61 (s, 3H, 2-NCH3), 3.27 (q, J = 7.5 Hz, 2H, 6-CH2CH3) 3.23 (s, 3H, 4-NCH3), 1.32 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (400 MHz DMSO6): Not obtained due to low solubility of the compound; HRMS m/z 435.1299 (Calculated for C22H19N4O6 [M + H]+: 435.1305); purified in column chromatography with ethyl acetate; yield: 12%.
The 6-ethyl-8-((4-fluorophenyl)amino)-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (26): Prepared from 3 and 4-fluoroaniline using general procedure C; burgundy red solid; mp 216.1–216.9 °C (d); 1H-NMR (400 MHz, CDCl3) δ7.49 (s, 1H, NH), 7.22 (dt, JH,H = 7.9, JF,H = 2.6 Hz, 2H, 2′-H and 6′-H), 7.13 (t, JH,H = 8.5, JF,H = 8.5 Hz, 2H, 3′-H and 5′-H), 6.30 (s, 1H, 9-H), 3.76 (s, 3H, 2-NCH3), 3.47 (s, 3H, 4-NCH3), 3.41 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.37 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 182.1, 179.9, 170.2, 160.4 (d, 1C, JF,C = 253.5 Hz, 4′), 158.8, 153.1, 151.2, 149.3, 145.1, 133.1(d, 1C, JF,C = 2.9 Hz, 1′), 124.7 (d, 2C, JF,C = 8.3 Hz, 2′ and 6′), 119.4, 116.8 (d, 2C, JF,C = 22.8 Hz, 3′ and 5′), 106.0, 103.3, 31.8, 30.2, 29.1, 12.1; HRMS m/z 409.1307 (Calculated for C21H18FN4O4 [M + H]+: 409.1312); purified in column chromatography with chloroform: ethyl acetate = 9:1; yield: 70%.
The 8-((4-chlorophenyl)amino)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (27); Prepared from 3 and 4-chloroaniline using general procedure C; purple solid; mp 206.0–207.0 °C (d); 1H-NMR (400 MHz, CDCl3) δ 7.56 (s,1H, NH), 7.39 (d, J = 8.8 Hz, 2H, 3′-H and 5′-H), 7.20 (d, J = 8.8 Hz, 2H, 2′-H and 6′-H), 6.40 (s, 1H, 9-H), 3.76 (s, 3H, 2-NCH3), 3.47 (s, 3H, 4-NCH3), 3.40 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.36 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 182.2, 179.8, 170.3, 158.6, 153.1, 151.2, 149.1, 144.3, 135.8, 131.0, 129.3 (2C), 123.5 (2C), 119.3, 105.9, 104.0, 31.8, 30.2, 29.1, 12.1; HRMS m/z 425.1021 (Calculated for C21H18ClN4O4 [M + H]+: 425.1017); purified in column chromatography with chloroform: ethyl acetate: petroleum ether = 2:1:2; yield: 53%.
The 8-((4-bromophenyl)amino)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (28): Prepared from 3 and 4-bromoaniline using general procedure C; red solid; mp > 250.0 °C; 1H-NMR (400 MHz, CDCl3) δ7.55 (s, 1H, NH), 7.54 (d, J = 8.7 Hz, 2H, 3′-H and 5′-H), 7.15 (d, J = 8.7 Hz, 2H, 2′-H and 6′-H), 6.42 (s, 1H, 9-H), 3.76 (s, 3H, 2-NCH3), 3.47 (s, 3H, 4-NCH3), 3.40 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.36 (t, J = 7.3 Hz, 3H, 6-CH2CH3);13C-NMR (100 MHz, CDCl3) δ 182.2, 179.8, 170.3, 158.6, 153.1, 151.2, 149.1, 144.2, 136.4, 132.9 (2C), 123.7 (2C), 119.3, 118.7, 105.9, 104.1, 31.8, 30.2, 29.1, 12.1; HRMS m/z 469.0515 (Calculated for C21H18BrN4O4 [M + H]+: 469.0511); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 4:1:4; yield: 67%.
The 6-ethyl-8-((4-iodophenyl)amino)-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (29): Prepared from 3 and 4-iodoaniline using general procedure C; purple solid; mp > 250 °C; 1H-NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.1 Hz, 3H, 3′-H and 5′-H), 7,55 (s, 1H, NH), 7.02 (d, J = 8.2 Hz, 2H, 2′-H and 6′-H), 6.43 (s, 1H, 9-H), 3.76 (s, 3H, 2-NCH3), 3.47 (s, 3H, 4-NCH3), 3.40 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.36 (t, J = 7.3 Hz, 3H, 6-CH2CH3);13C-NMR (100 MHz, CDCl3) δ 182.2, 179.8, 170.3, 158.6, 153.1, 151.2, 149.4, 144.0, 138.8 (2C), 137.1, 123.8 (2C), 119.3, 105.9, 104.3, 89.3, 31.8, 30.2, 29.1, 12.0; HRMS m/z 517.0372 (Calculated for C21H18IN4O4 [M + H]+: 517.0373); purified in column chromatography with chloroform; yield: 94%.
The 6-ethyl-8-(ethylthio)-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (30); Prepared from 3 and ethanethiol; orange solid; mp 171.3–172.8 °C; 1H-NMR (400 MHz, CDCl3) δ 6.67 (s, 1H, 9-H), 3.75 (s, 3H, 2-NCH3), 3.46 (s, 3H, 4-NCH3), 3.37 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 2.85 (q, J = 7.4 Hz, 2H, 8-S-CH2CH3), 1.43 (t, J = 7.4 Hz, 3H, 8-S-CH2CH3), 1.33 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 180.8, 180.7, 170.7, 158.5, 155.1, 152.7, 151.1, 147.1, 126.5, 120.8, 105.4, 31.7, 30.2, 29.1, 24.9, 12.5 12.1; HRMS m/z 360.1010 (Calculated for C17H18N3O4S [M + H]+: 360.1018); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 15:3:4; yield: 48%.
The 6-ethyl-2,4-dimethyl-8-(propylthio)pyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (31): Prepared from 3 and propane-1-thiol using general procedure B; orange solid; mp 163.8–164.9 °C; 1H-NMR (400 MHz, CDCl3) δ 6.68 (s, 1H, 9-H), 3.76 (s, 3H, 2-NCH3), 3.48 (s, 3H, 4-NCH3), 3.39 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 2.81 (t, J =7.3 Hz, 2H, 8-S-CH2CH2CH3), 1.82 (m, 2H, 8-S-CH2CH2CH3), 1.35 (t, J =7.3 Hz, 3H, 6-CH2CH3), 1.11 (t, J =7.4 Hz, 3H, 8-S-CH2CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 180.84, 180.78, 170.7, 158.6, 155.4, 152.7, 151.1, 147.2, 126.5, 120,8, 105.4, 32.8, 31.7, 30.2, 29.1, 20.9, 13.7, 12.1; HRMS m/z 374.1172 (Calculated for C18H20N3O4S [M + H]+: 374.1175); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 2:1:4; yield: 40%.
The 8-(butylthio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (32): Prepared from 3 and butane-1-thiol using general procedure B; orange solid; mp 158.8–160.5 °C; 1H-NMR (400 MHz, CDCl3) δ 6.67 (s, 1H, 9-H), 3.75 (s, 3H, 2-NCH3), 3.46 (s, 3H, 4-NCH3), 3.37 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 2.82 (t, J = 7.4 Hz, 2H, 8-S-CH2CH2CH2CH3), 1.75 (dt, J = 15.0, J = 7.4 Hz, 2H, 8-S-CH2CH2CH2CH3),1.51 (dq, J = 14.6, J = 7.3 Hz, 2H, 8-CH2CH2CH2CH3), 1.34 (t, J = 7.3 Hz, 3H, 6-CH2CH3), 0.97 (t, J = 7.4 Hz, 3H, 8-CH2CH2CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 180.82, 180.75, 170.7, 158.5, 155.4, 152.7, 151.1, 147.1, 126.4, 120.8, 105.4, 31.7, 30.6, 30.2, 29.3, 29.1, 22.2, 13.5, 12.1; HRMS m/z 388.1326 (Calculated for C19H22N3O4S [M + H]+: 388.1331); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 4:0.5:3; yield: 58%.
The 8-pentylthio-6-ethyl-2,4-dimethyl-pyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (33): Prepared from 3 and pentane-1-thiol using general procedure B; orange solid; mp 158.6–160.4 °C; 1H-NMR (400 MHz, CDCl3) δ 6.67 (s, 1H, 9-H), 3.75 (s, 3H, 2-NCH3), 3.46 (s, 3H, 4-NCH3), 3.37 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 2.81 (t, J = 7.3 Hz, 2H, 8-S-CH2CH2CH2CH2CH3), 1.77 (dt, J = 7.5, J = 7.4 Hz, 2H, 8-S-CH2CH2CH2CH2CH3), 1.46 (dt, J = 14.2, J = 6.9 Hz, 2H, 8-S-CH2CH2CH2CH2CH3), 1.37–1.28 (m, 5H, 6-CH2CH3 and 8-S-CH2CH2CH2CH2CH3), 0.97 (t, J = 7.4 Hz, 3H, 8-S-CH2CH2CH2CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 180.95, 180.86, 170.81, 158.64, 155.54, 152.78, 151.23, 147.24, 126.54, 120.93, 105.51, 31.81, 31.26, 30.93, 30.27, 29.17, 27.14, 22.28, 13.99, 12.22; HRMS m/z 402.1483 (Calculated for C20H24N3O4S [M + H]+: 402.1488); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 12:1:9; yield: 43%.
The 8-hexylthio-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (34): Prepared from 3 and hexane-1-thiol using general procedure B; orange solid; mp 134.6–137.0 °C; 1H-NMR (400 MHz, CDCl3) δ 6.65 (s, 1H, 9-H), 3.73 (s, 3H, 2-NCH3), 3.44 (s, 3H, 4-NCH3), 3.35 (q, J = 7,3 Hz, 2H, 6-CH2CH3), 2.80 (t, J = 7.3 Hz, 2H, 8-S-CH2CH2CH2CH2CH2CH3), 1.74 (q, J = 7.3 Hz, 2H, 8-S-CH2CH2CH2CH2CH2CH3), 1.47 (q, 2H, 8-CH2CH2CH2CH2CH2CH3), 1.47 (m, 4H, 8-CH2CH2CH2CH2CH2CH3 and 8-CH2CH2CH2CH2CH2CH3), 1.32 (t, J = 7.3 Hz, 3H, 6-CH2CH3), 0.88 (t, J = 7.3 Hz, 3H, 8-CH2CH2CH2CH2CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 180.8, 180.7, 170.7, 158.4, 155.4, 152.5, 151.1, 147.1, 126.4, 120.8, 105.4, 31.7 31.2, 30.8, 30.1, 29.0, 28.7, 27.3, 22.5, 14.0, 12.1; HRMS m/z 402.1483 (Calculated for C20H24N3O4S [M + H]+: 402.1488); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 2:1:4; yield: 52%.
The 6-ethyl-2,4-dimethyl-8,9-bis(phenylthio)pyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (35): Prepared from 3 and benzenethiol using general procedure D; red solid; mp 188.9–191.5 °C; 1H-NMR (400 MHz, CDCl3) δ 7.58–7.54 (m, 2H, 8-C6H5 or 9-C6H5), 7.43–7.37 (m, 5H, 8-C6H5 or 9-C6H5), 7.34–7.27 (m, 3H, 8-C6H5 or 9-C6H5), 3.71 (s, 3H, 2-NCH3), 3.31 (s, 3H, 4-NCH3), 3.06 (q, J = 7.4 Hz, 2H, 6-CH2CH3), 1.13 (q, J = 7.4 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 179.3, 176.8, 169.9, 157.6, 152.1, 151.1, 150.5, 147.7, 143.7, 133.3, 133.2 (2C), 131.2 (2C), 130.2, 129.4 (2C), 129.3 (2C), 128.9, 127.9, 122.1, 104.8, 31.0, 30.1, 28.8, 12.3; HRMS m/z 516.1058 (Calculated for C27H22N3O4S2 [M + H]+: 516.1052); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 4:1:5; yield: 39%.
The 8,9-bis(4-chlorophenylthio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (36): Prepared from 3 and 4-chlorobenzenethiol using general procedure D; brown solid; mp 207.8–209.8 °C; 1H-NMR (400 MHz, CDCl3) δ 7.49 (d, J = 8.5 Hz, 2H, 2″-H and 6″-H), 7.36 (d, J = 8.6 Hz, 2H, 3″-H and 5″-H), 7.35 (d, J = 8.7 Hz, 2H, 2′-H and 6′-H), 7.29 (d, J = 8.5 Hz, 2H, 3′-H and 5′-H), 3.71 (s, 3H, 2-NCH3), 3.33 (s, 3H, 4-NCH3), 3.10 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.17 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 179.4, 176.40, 170.1, 157.6, 152.1, 151.2, 151.0, 147.8, 142.3, 135.7, 134.8 (2C), 134.3, 132.6 (2C), 131.4, 129.7 (2C), 129.5 (2C), 128.2, 121.7, 104.8, 31.2, 30.1, 28.8, 12.3; HRMS m/z 584.0263 (Calculated for C27H20Cl2N3O4S2 [M + H]+: 548.0272); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 1:1:3; yield: 49%.
The 8,9-bis(propylthio)-6-ethyl-2,4-dimethyl-pyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (37): Prepared from 3 and propane-1-thiol using general procedure D; red solid; mp 138.9–140.2 °C; 1H-NMR (400 MHz, CDCl3) δ 3.75 (s, 3H, 2-NCH3), 3.46 (s, 3H, 4-NCH3), 3.34 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 3.27 (t, J = 7.2 Hz, 2H, 9-CH2CH2CH3), 3.08 (t, J = 7.3 Hz, 2H, 8-CH2CH2CH3), 1.76 (h, 2H, 9-CH2CH2CH3), 1.60 (h, 2H, 8-CH2CH2CH3), 1.33 (t, J = 7.3 Hz, 3H, 6-CH2CH3), 1.07 (t, J = 7.3 Hz, 3H, 9-CH2CH2CH3), 1.00 (t, J = 7.3 Hz, 3H, 8-CH2CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 180.4, 176.6, 169.8, 158.6, 152.4, 151.8, 151.1, 148.7, 142.2, 122.0, 104.6, 36.0, 35.1, 31.2, 30.1, 28.9, 24.2, 23.9, 13.3, 13.1, 12.5; HRMS m/z 448.1365 (Calculated for C21H26N3O4S2 [M + H]+: 448.1365); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 5:1:14; yield: 86%.
The 8-((2-bromo-4-chlorophenyl)thio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (38); Prepared from 3 and 2-bromo-4-chlorobenzenethiol using general procedure B; orange solid; mp 198.4–200.2 °C; 1H-NMR (400 MHz, CDCl3) δ 7.82 (d, J = 1.9 Hz, 1H, 3′-H), 7.59 (d, J = 8.3 Hz, 1H, 6′-H), 7.44 (dd, J = 8.3, J = 1.9 Hz, 1H, 5′-H), 6.06 (s, 1H, 9-H), 3,76 (s, 3H, 2-NCH3), 3,44 (s, 3H, 4-NCH3), 3.42 (q, J = 7.2 Hz, 2H, 6-CH2CH3), 1.37 (t, J = 7.2 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 181.0, 180.5, 170.8, 158.3, 153.4, 152.8, 151.0, 147.1, 138.5, 138.2, 132.3, 131.2, 129.5, 128.0, 127.3, 120.5, 105.5, 31.7, 30.2, 29.1, 12.1; HRMS m/z 519.9739 (Calculated for C21H16BrClN3O4S [M + H]+: 519.9733); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 1:5:4; yield: 70%.
The 8-((2,6-dimethoxyphenyl)thio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (39): Prepared from 3 and 2,6-dimethoxybenzenethiol using general procedure B; red solid; mp 223.0–223.7 °C (d); 1H-NMR (400 MHz, CDCl3) δ 7.45 (t, J = 8.4 Hz, 1H, 4′-H), 6.66 (d, 2H, 3′-H and 5′-H), 6.07 (s, 1H, 9-H), 3.84 (s, 6H, 2′-OCH3 and 6′-OCH3), 3.74 (s, 3H, 2-NCH3), 3.43 (s, 3H, 4-NCH3), 3.41 (q, J = 7.2 Hz, 2H, 6-CH2CH3), 1.36 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 181.3 (2C), 170.5, 161.3 (2C), 158.6, 154.1, 152.6, 151.1, 147.5, 133.2, 126.8, 121.0, 105.4, 104.5 (2C), 102.0, 56.4 (2C), 31.7, 30.2, 29.0, 12.2; HRMS m/z 468.1226 (Calculated for C23H22N3O6S [M + H]+: 468.1229); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 9:1:3; yield: 63%.
The 8-((5-bromo-2-methoxyphenyl)thio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (40): Prepared from 3 and 5-bromo-2-methoxybenzenethiol using general procedure B; orange solid; mp 221.0–222.0 °C (d); 1H-NMR (400 MHz, CDCl3) δ 7.63 (q, J = 2.4 Hz, 1H, 6′-H), 7.61 (d, J = 8.7, J = 2.6 Hz, 1H, 4′-H), 6.93 (d, J = 8.6 Hz, 1H, 3′-H), 6.11 (s, 1H, 9-H), 3.85 (s, 3H, 2′-OCH3), 3.75 (s, 3H, 2-NCH3), 3.44 (s, 3H, 4-NCH3), 3.41 (q, J = 7.2 Hz, 2H, 6-CH2CH3), 1.36 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 181.2, 180.8, 170.7, 159.2, 158.4, 153.8, 152.7, 151.1, 147.2, 139.5, 135.7, 127.7, 120.7, 116.7, 113.5, 113.3, 105.4, 56.4, 31.7, 30.2, 29.1, 12.1; HRMS m/z 516.0237 (Calculated for C22H19BrN3O5S [M + H]+: 516.0229); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 20:1:4; yield: 76%.
The 8-((3,5-dichlorophenyl)thio)-6-ethyl-2,4-dimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (41): Prepared from 3 and 3,5-dichlorobenzenethiol using general procedure B; yellow solid; mp 179.8–182.0 °C; 1H-NMR (400 MHz, CDCl3) δ 7.44 (s, 2H, 2′-H and 6′-H), 7.52 (s, 1H, 4′-H); 6.24 (s, 1H, 9-H), 3.75 (s, 3H, 2-NCH3), 3.43 (s, 3H, 4-NCH3), 3.40 (q, J = 7.3 Hz, 2H, 6-CH2CH3), 1.36 (t, J = 7.3 Hz, 3H, 6-CH2CH3); 13C-NMR (100 MHz, CDCl3) δ 181.1, 180.3, 170.9, 158.6, 145.9, 152.8, 151.0, 147.0, 136.6, 133.6 (2C), 131.1, 130.5, 128.4 (2C), 120.3, 105.5, 31.7, 30.2, 29.1, 12.1; HRMS m/z 476.0235 (Calculated for C21H16Cl2N3O4S [M + H]+: 476.0239); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 20:1:7; yield: 69%.
The 8-phenylthio-2,4,6-trimethyl-pyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (42): Prepared from 4 and benzenethiol using general procedure B; yellow solid; mp 206.0–208.0 °C (d); 1H-NMR (400 MHz, CDCl3) δ 7.53 (m. 5H, 2′-H, 3′-H, 4′-H, 5′-H and 6′-H), 6.18 (s, 1H, 9-H), 3.74 (s, 3H, 2-NCH3), 3.43 (s, 3H, 4-NCH3), 3.01 (s, 3H, 6-CH3); 13C-NMR (100 MHz, CDCl3) δ 181.1, 180.9, 166.3, 158.3, 156.5, 152.7, 151.0, 146.9, 135.7 (2C), 130.7, 130.5 (2C), 128.1, 127.1, 120.9, 105.7, 30.2, 29.1, 26.9; HRMS m/z 394.0862 (Calculated for C20H16N3O4S [M + H]+: 394.0862); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 12:1:8; yield: 65%.
The 8-(4-methoxy-phenylthio)-2,4,6-trimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (43): Prepared from 4 and 4-methoxybenzenethiol using general procedure B; orange solid; mp 198.0–199.0 °C (d); 1H-NMR (400 MHz, CDCl3) δ 7.43 (d. 2H, 3′-H and 5′-H), 7.02 (d. 2H, 2′-H and 6′-H), 6.16 (s, 1H, 9-H), 3.86 (s, 3H, 4′-OCH3), 3.73 (s, 3H, 2-NCH3), 3.43 (s, 3H, 4-NCH3), 3.00 (s, 3H, 6-CH3); 13C-NMR (100 MHz, CDCl3) δ 181.2, 181.0, 166.2, 161.6, 158.3, 157.3, 152.6, 151.0, 147.0, 137.2 (2C), 128.0, 121.0, 117.2, 116.1 (2C), 105.7, 55.5, 30.2, 29.0, 26.9; HRMS m/z 424.0963 (Calculated for C21H18N3O5S [M + H]+: 424.0967); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 3:1:4; yield: 69%.
The 8-((4-fluorophenyl)thio)-2,4,6-trimethylpyrimido[4,5-c]isoquinoline-1,3,7,10(2H,4H)-tetraone (44): Prepared from 4 and 4-fluorobenzenethiol using general procedure B; yellow solid; mp 211.0–212.0 °C (d); 1H-NMR (400 MHz, CDCl3) δ 7.52 (dd, JH,H = 8.7, JH,H = 5.2 Hz, 2H, 2′-H and 6′-H), 7.21 (t, JH,H = 8.5, JF,H = 8.5 Hz, 2H, 3′-H and 5′-H), 6.15 (s, 1H, 9-H), 3.73 (s, 3H, 2-NCH3), 3.43 (s, 3H, 4-NCH3), 3.01 (s, 3H, 6-CH3); 13C-NMR (100 MHz, CDCl3) δ 181.2, 180.9, 166.4, 164.4 (d, 1C, JF,C = 252.9 Hz, 4′), 158.4, 156.4, 152.8, 151.1, 146.9, 138.0 (d, 2C, JF,C = 8.8 Hz, 2′ and 6′), 128.2, 122.5 (d, 1C, JF,C = 3.6 Hz, 1′), 121.0, 118.0 (d, 2C, JF,C = 22.1 Hz, 3′ and 5′), 105.8, 30.4, 29.2, 27.0; HRMS m/z 412.0771 (Calculated for C20H15FN3O4S [M + H]+: 412.0767); purified in column chromatography with dichloromethane: ethyl acetate: petroleum ether = 1:1:2; yield: 69%.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



























































