
Effect of Indole-Containing Pyrazino[2,1-b]quinazoline-3,6-diones in the Virulence of Resistant Bacteria
 , , , , , , ,
, , , , , , ,  and
and 
Abstract
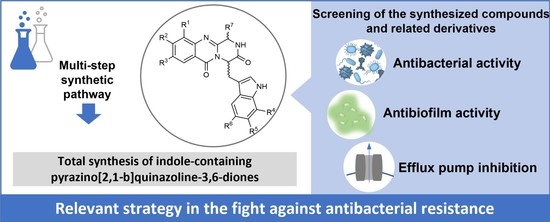
:
1. Introduction
2. Results and Discussion
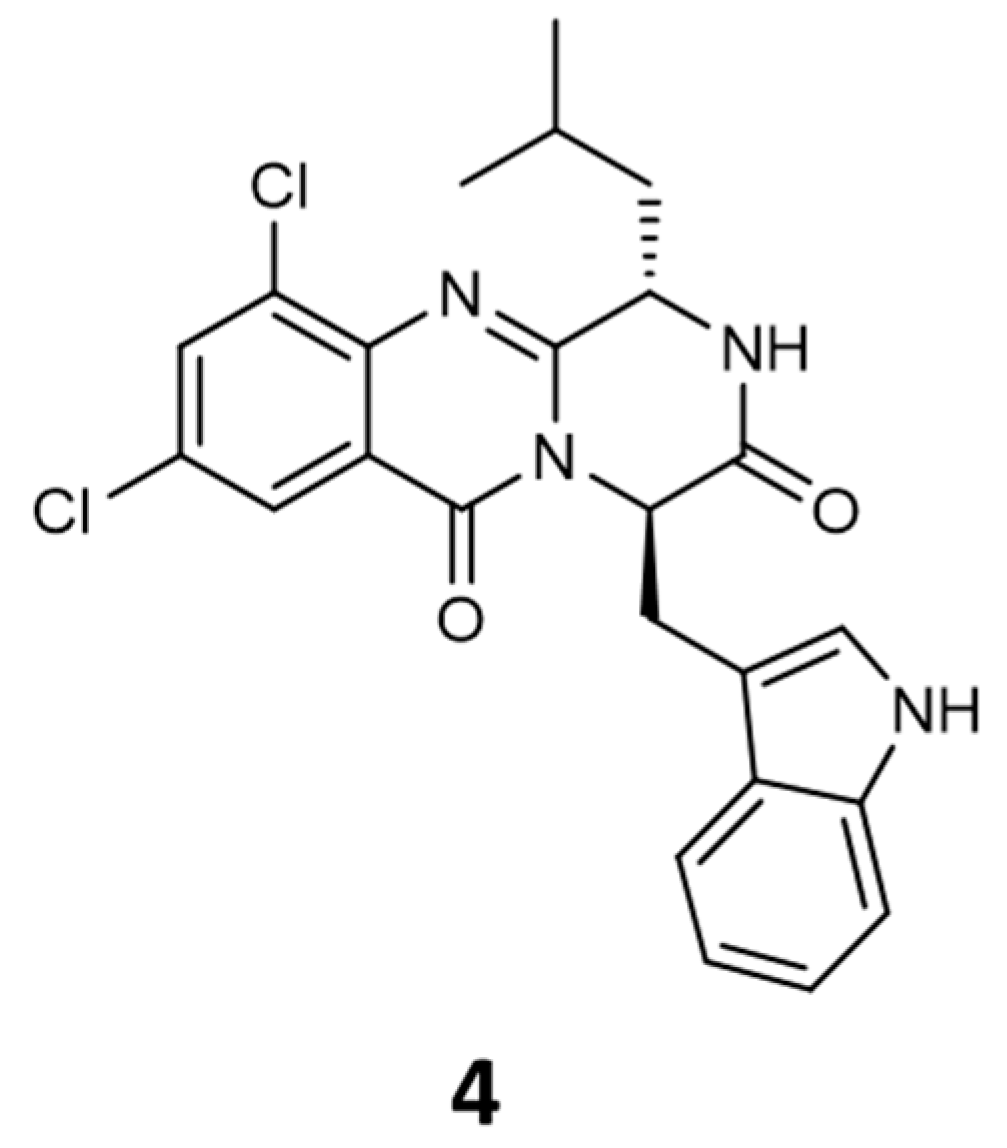
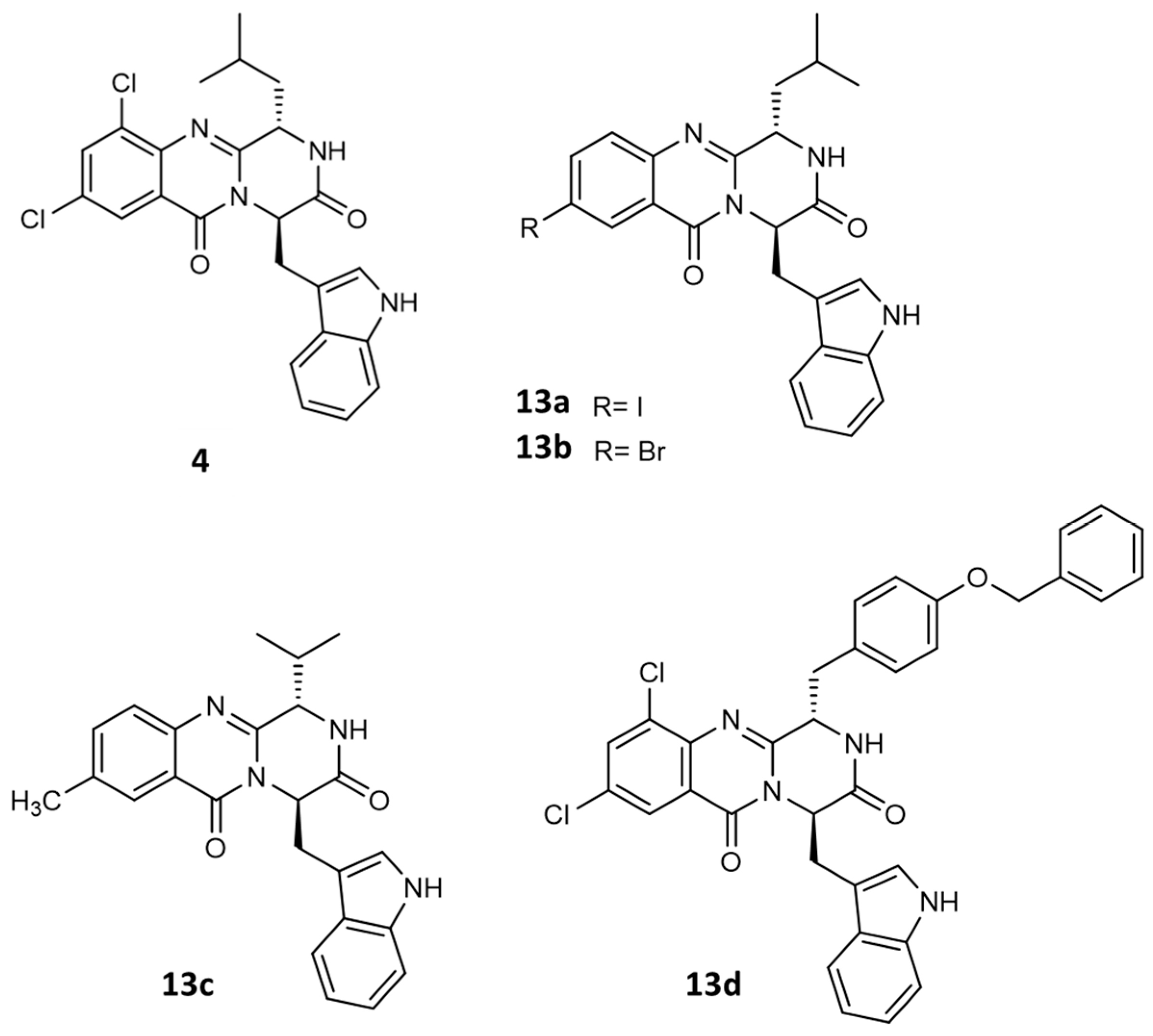
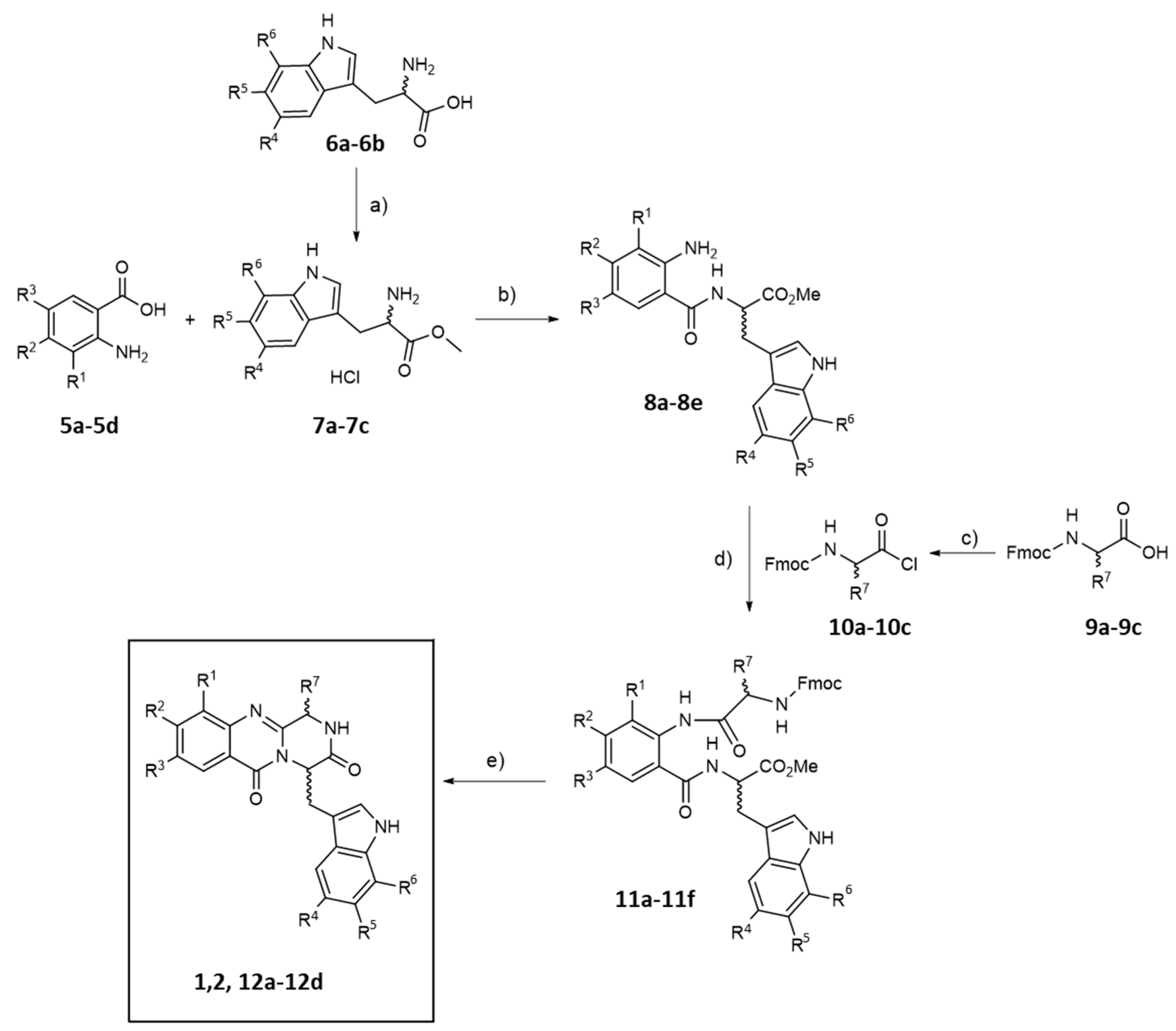
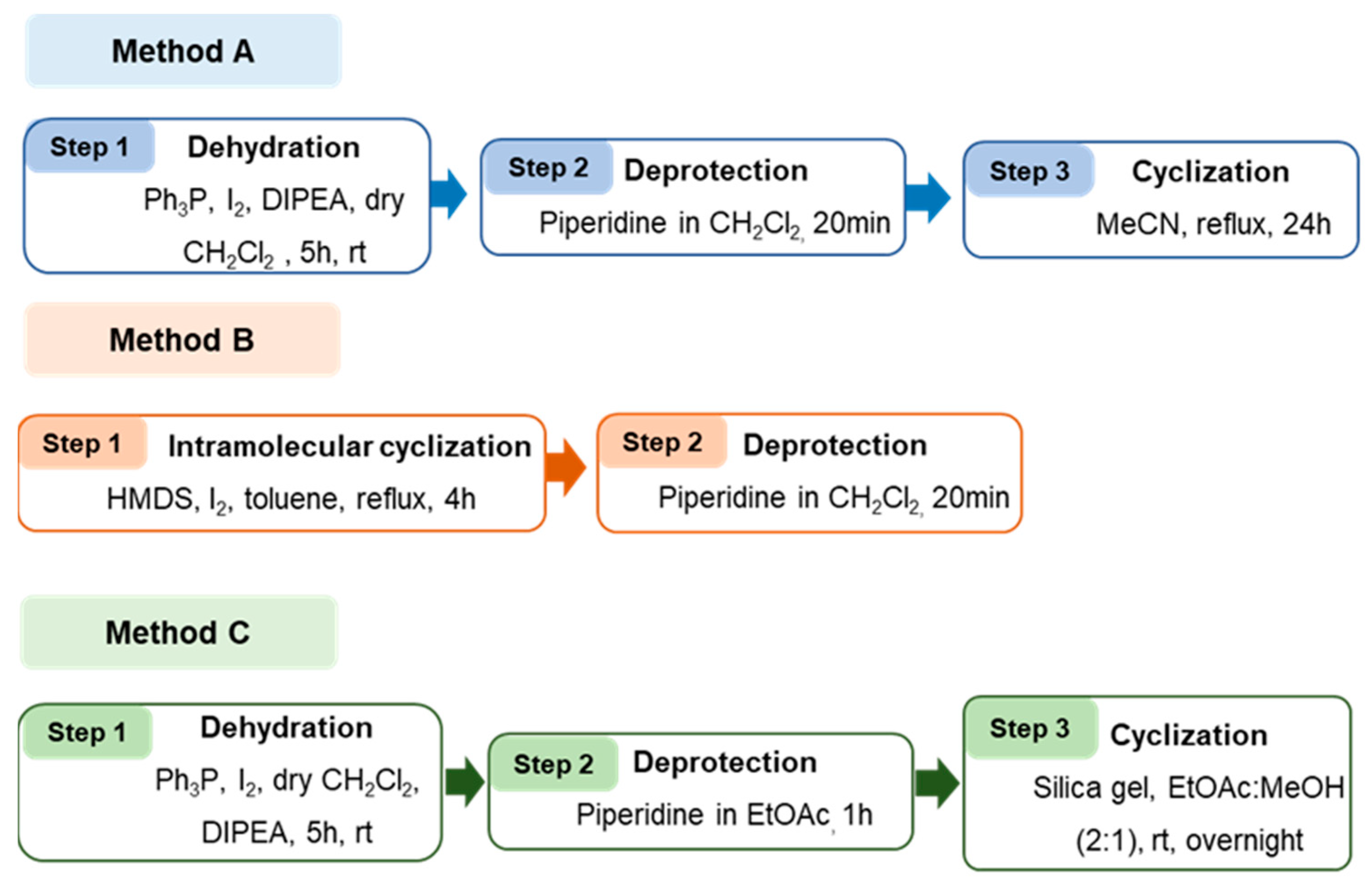
2.1. Synthesis and Structure Elucidation
2.2. Evaluation of Biological Activities
2.2.1. Antibacterial Activity
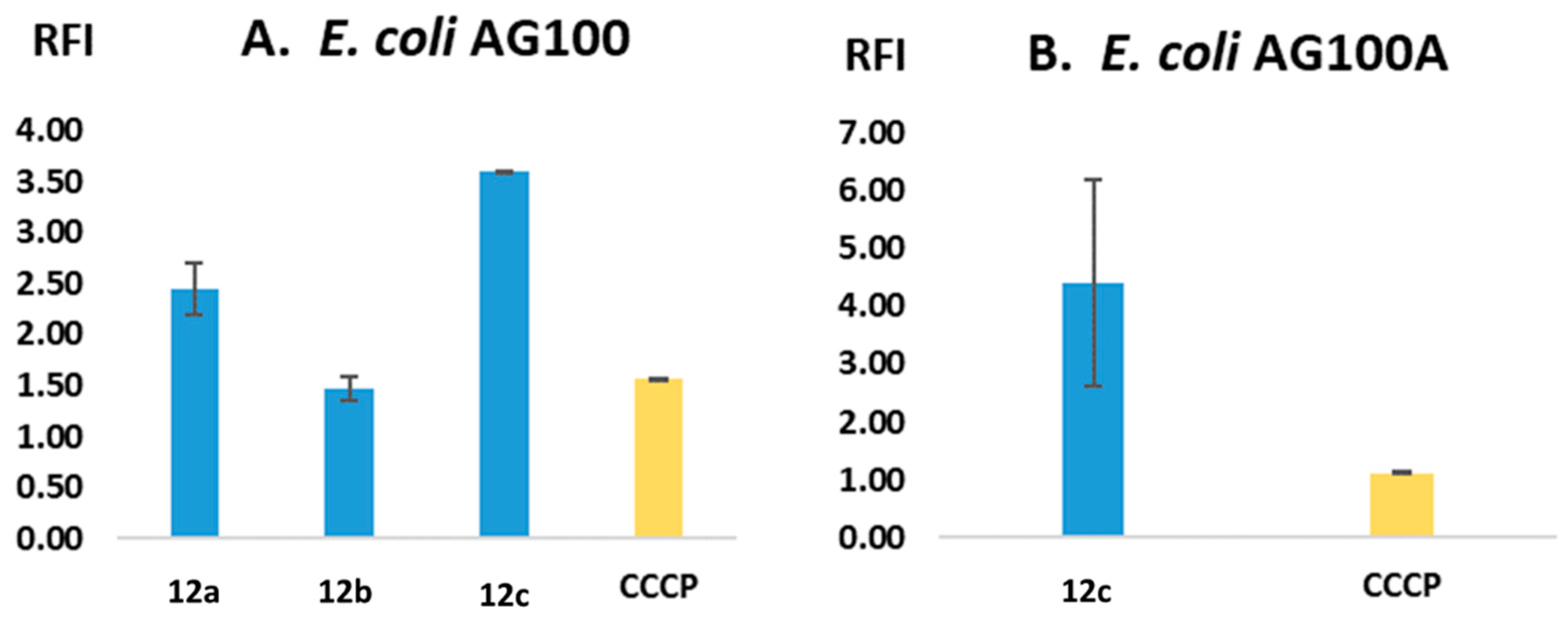
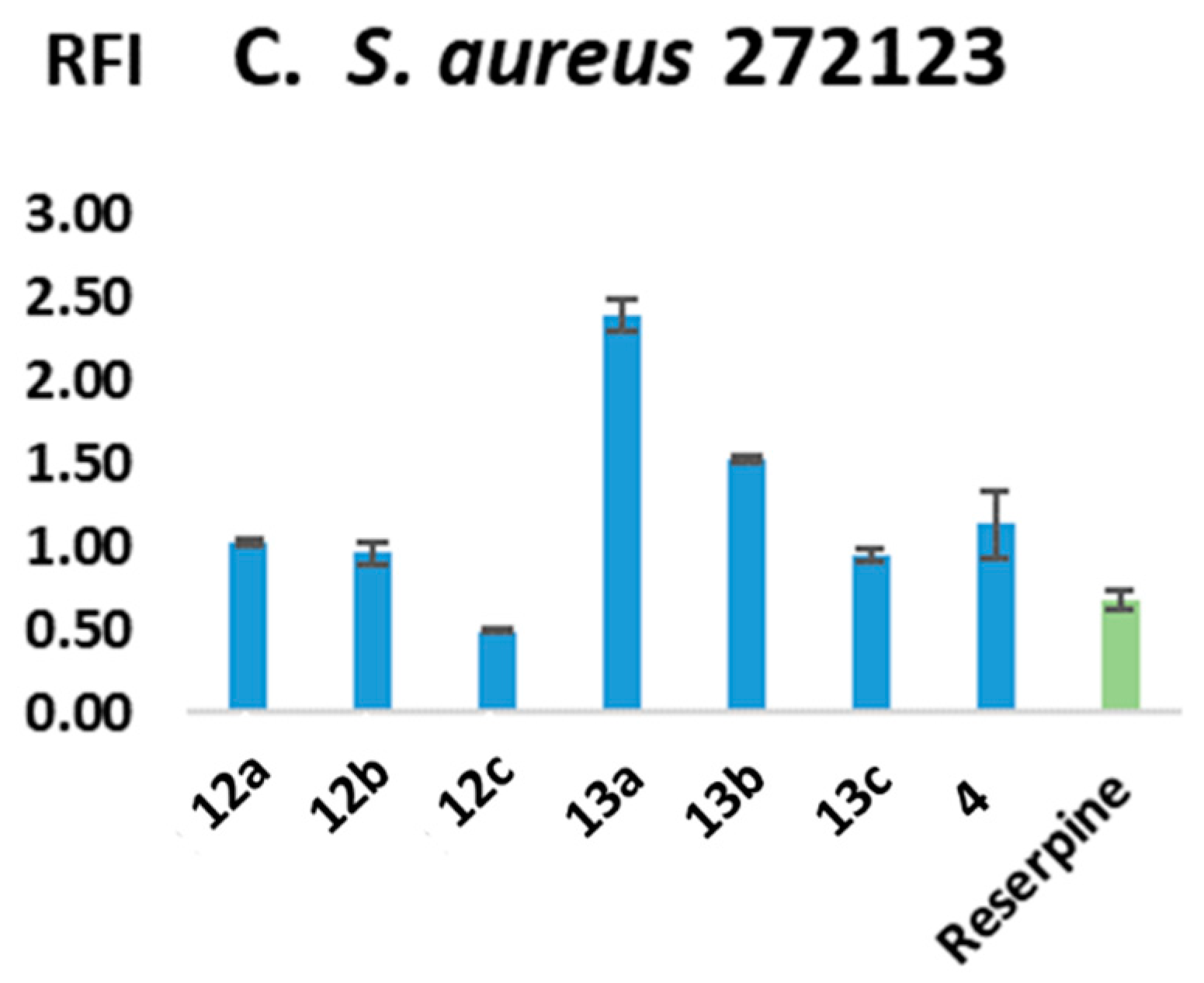
2.2.2. Efflux-Pump-Inhibiting (EPI) Capacity
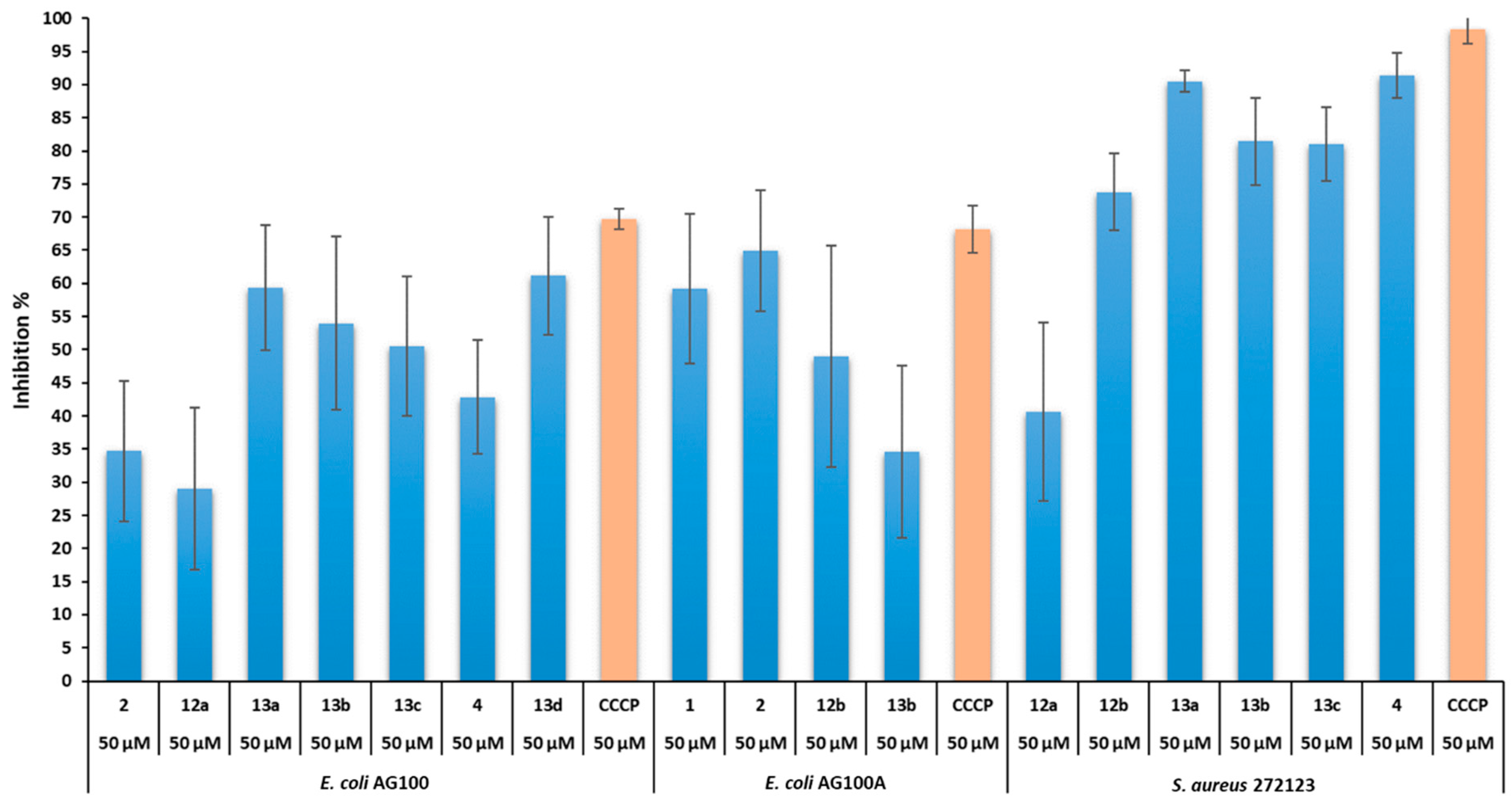
2.2.3. Antibiofilm Activity
2.2.4. Preliminary Antibiotic Potentiation Study
2.2.5. Cytotoxicity Assays
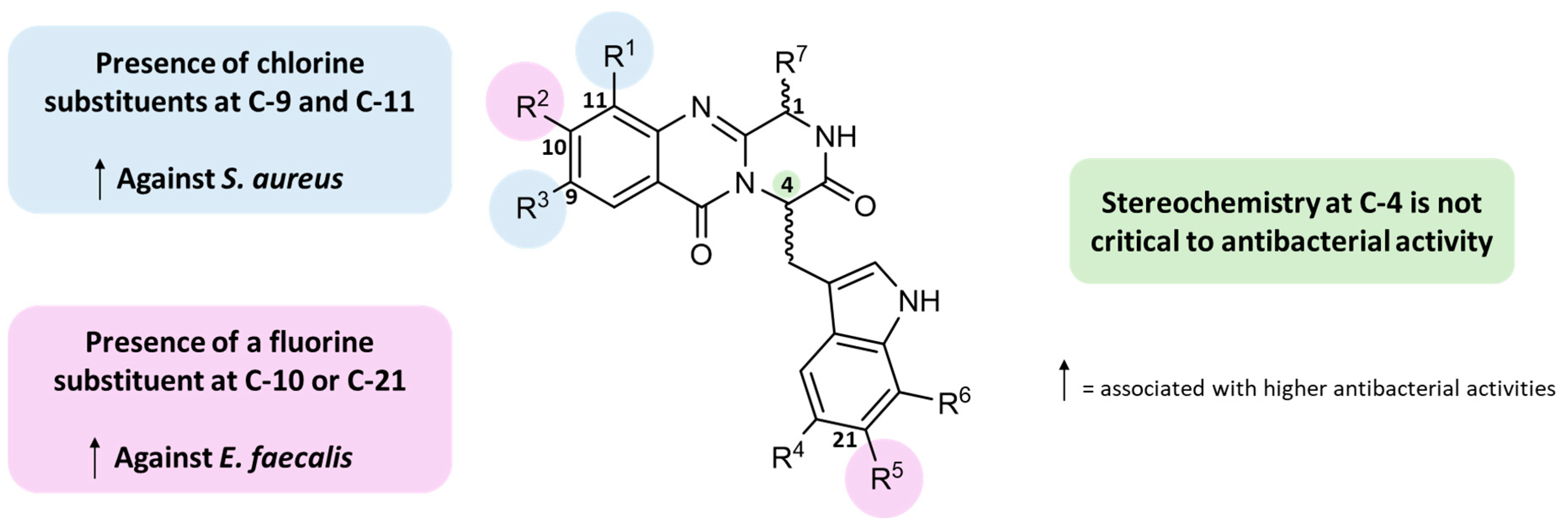
2.2.6. Structure–Activity Relationship (SAR)
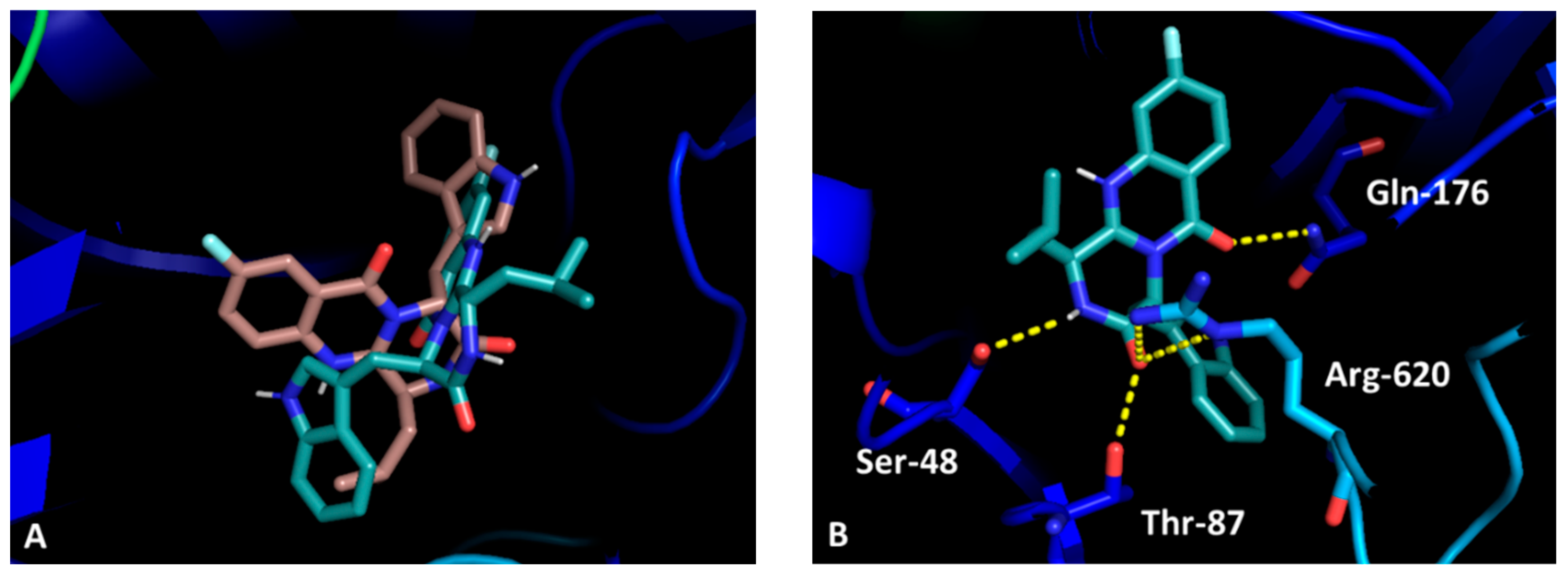
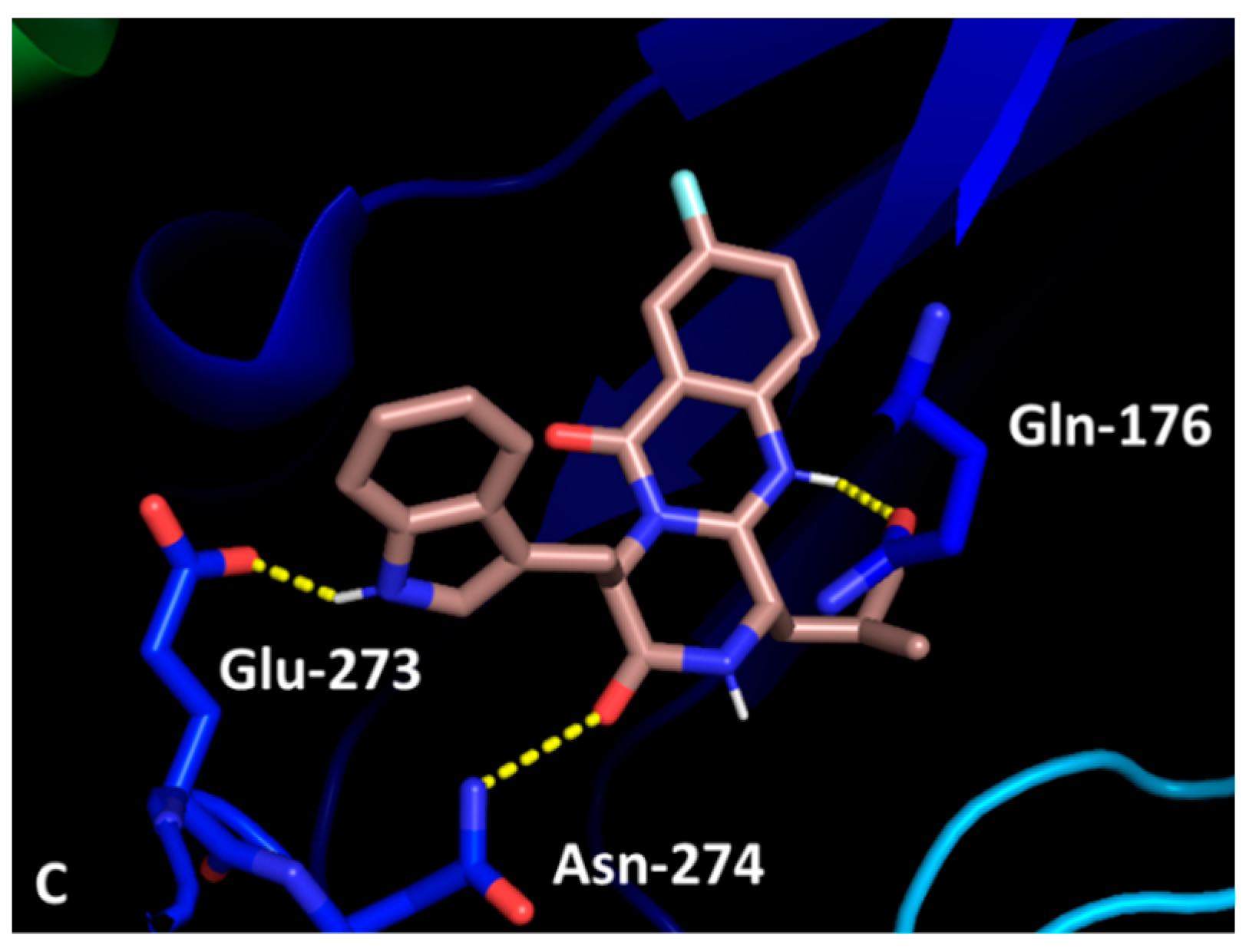
2.2.7. Docking Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. Materials and Methods
3.1.2. Synthesis of Tryptophan Methyl Esters Hydrochlorides 7b–7c
General Procedure
Synthesis of Methyl (S)-2-Amino-3-(6-fluoro-1H-indol-3-yl)propanoate (7b)
Synthesis of Methyl (S)-2-Amino-3-(7-chloro-1H-indol-3-yl)propanoate (7c)
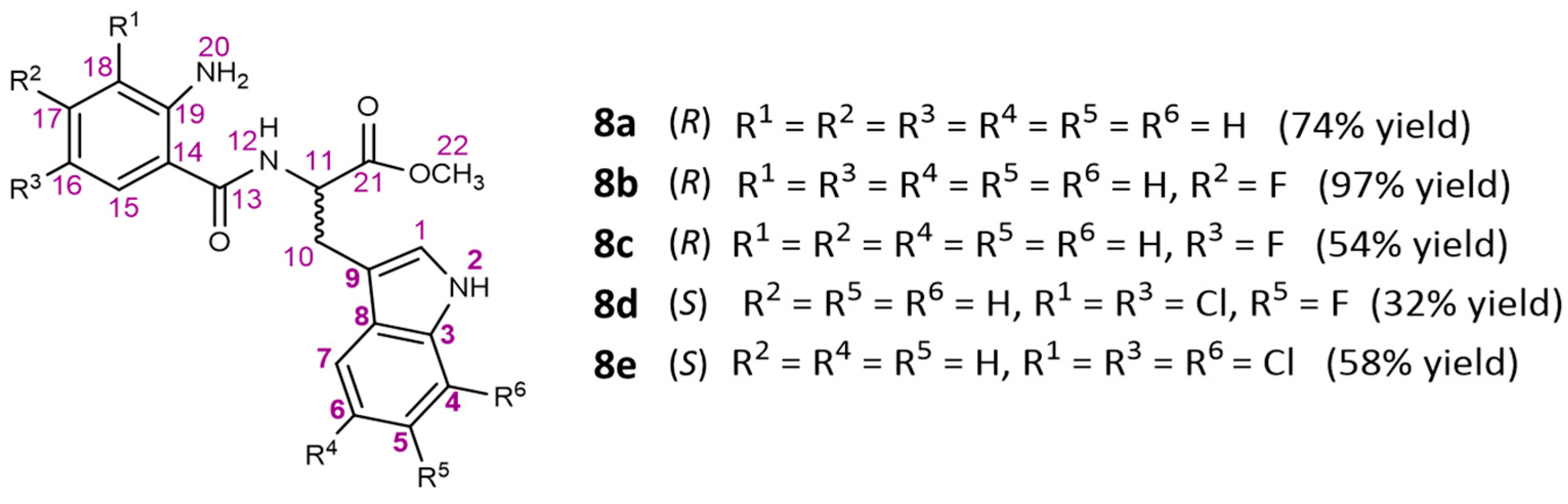
3.1.3. Synthesis of Linear Dipeptides 8a–8e
General Procedure
Synthesis of Methyl (2-Aminobenzoyl)-D-tryptophanate (8a)
Synthesis of Dipeptide Methyl (2-Amino-4-fluorobenzoyl)-D-tryptophanate (8b)
Synthesis of Dipeptide Methyl (2-Amino-5-fluorobenzoyl)-D-tryptophanate (8c)
Synthesis of Dipeptide Methyl (S)-2-(2-Amino-3,5-dichlorobenzamido)-3-(6-fluoro-1H-indol-3-yl)propanoate (8d)
Synthesis of Dipeptide Methyl (S)-2-(2-Amino-3,5-dichlorobenzamido)-3-(7-chloro-1H-indol-3-yl)propanoate (8e)
3.1.4. Synthesis of Acid Chlorides 10a–10c
General Procedure
Synthesis of Fmoc-Gly-Cl (10a)
Synthesis of Fmoc-L-Ala-Cl (10b)
Synthesis of Fmoc-L-Leu-Cl (10c)
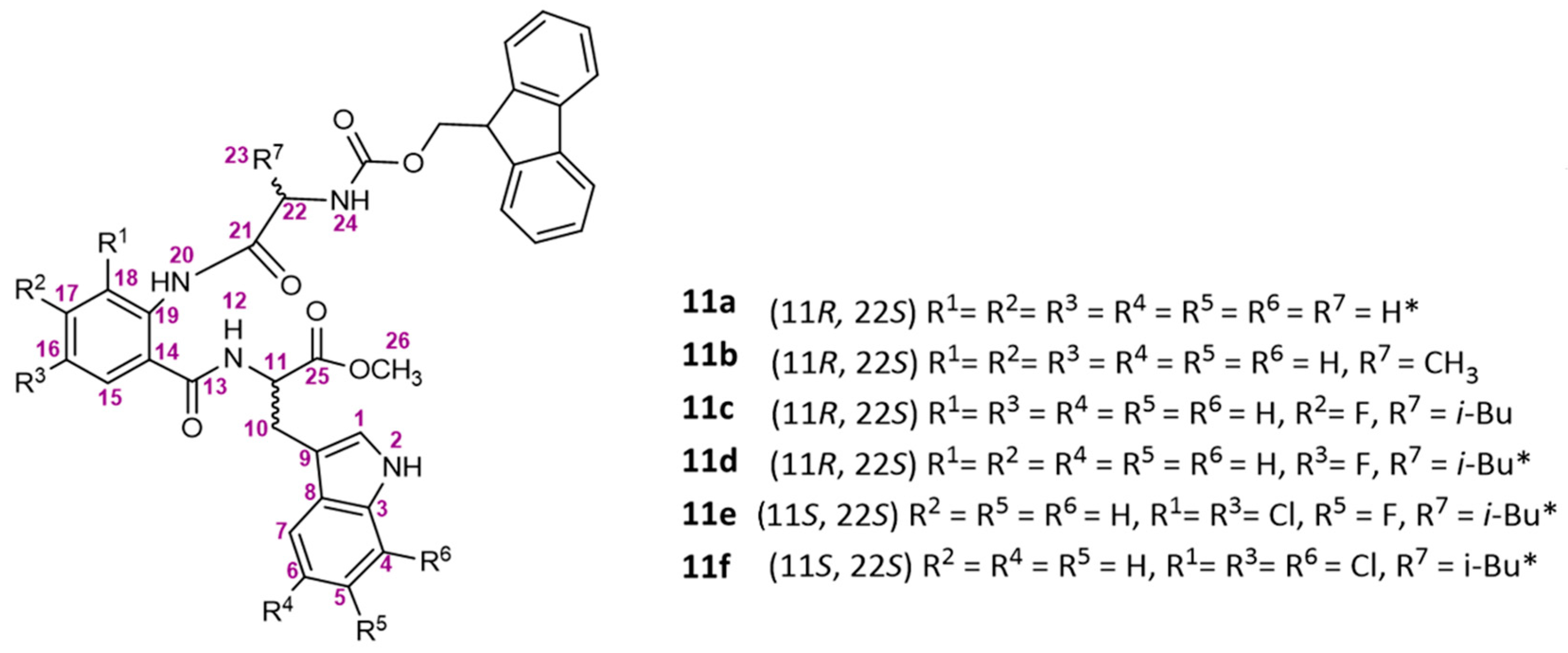
3.1.5. Synthesis of Linear Tripeptides 11a–11f
General Procedure
Synthesis of Methyl (2-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino) acetamido)benzoyl)-D-tryptophanate (11a)
Synthesis of Methyl (2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)propanamido)benzoyl)-D-tryptophanate (11b)
Synthesis of Methyl (2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentanamido)-4-fluorobenzoyl)-D-tryptophanate (11c)
Synthesis of Methyl (2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentanamido)-5-fluorobenzoyl)-D-tryptophanate (11d)
Synthesis of Methyl (S)-2-(2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentanamido)-3,5-dichlorobenzamido)-3-(6-fluoro-1H-indol-3-yl)propanoate (11e)
Synthesis of Tripeptide Methyl (S)-2-(2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentanamido)-3,5-dichlorobenzamido)-3-(7-chloro-1H-indol-3-yl)propanoate (11f)
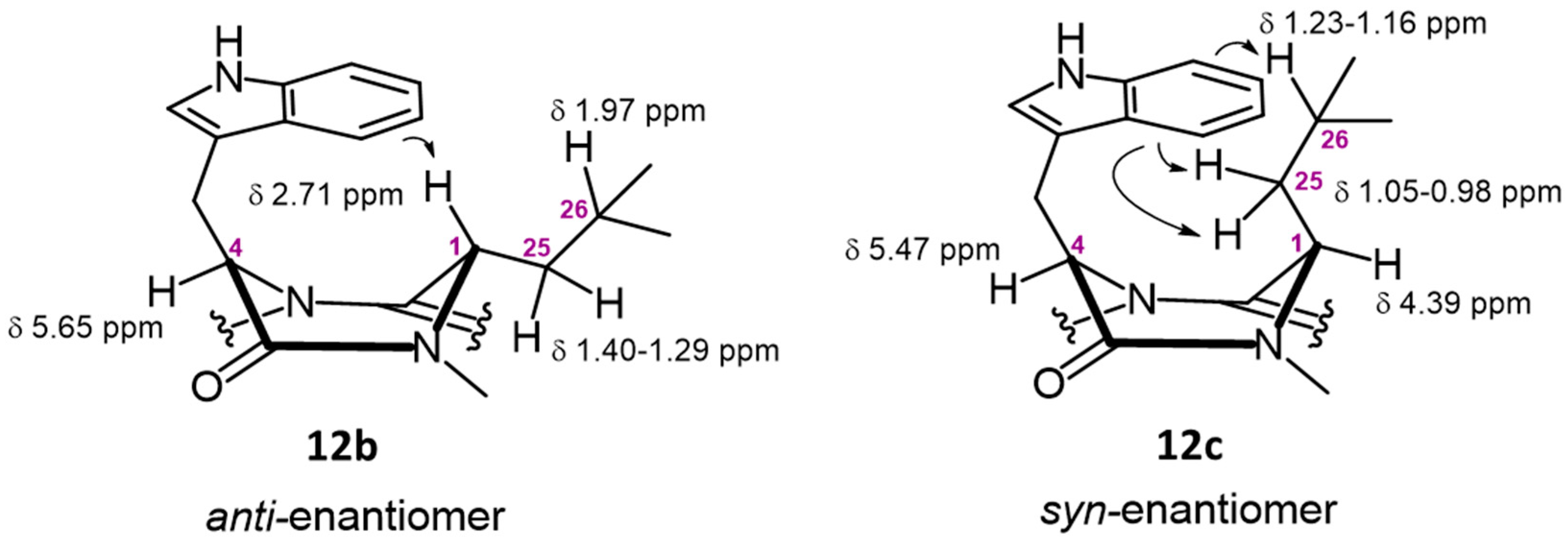
3.1.6. Synthesis of Derivatives 1, 2 and 12a–12d
Synthesis of (R)-4-((1H-Indol-3-yl)methyl)-1,2-dihydro-6H-pyrazino[2,1-b]quinazoline-3,6(4H)-dione (1, Glyantrypine)––Method A
General Procedure––Method C
Synthesis of (1S,4R)-4-((1H-Indol-3-yl)methyl)-1-methyl-1,2-dihydro-6H-pyrazino[2,1-b]quinazoline-3,6(4H)-dione (2, Fumiquinazoline F)
Synthesis of (1S,4R)-4-((1H-Indol-3-yl)methyl)-8-fluoro-1-isobutyl-1,2-dihydro-6H-pyrazino[2,1-b]quinazoline-3,6(4H)-dione (12a)
Synthesis of Alkaloid (1S,4R)-4-((1H-Indol-3-yl)methyl)-9-fluoro-1-isobutyl-1,2-dihydro-6H-pyrazino[2,1-b]quinazoline-3,6(4H)-dione (12b)
Synthesis of Alkaloid ((1S,4S)-8,10-Dichloro-4-((6-fluoro-1H-indol-3-yl)methyl)-1-isobutyl-1,2-dihydro-6H-pyrazino[2,1-b]quinazoline-3,6(4H)-dione (12c)
Synthesis of Alkaloid (1S,4S)-8,10-Dichloro-4-((7-chloro-1H-indol-3-yl)methyl)-1-isobutyl-1,2-dihydro-6H-pyrazino[2,1-b]quinazoline-3,6(4H)-dione (12d)
3.2. Microbiology
3.2.1. Reagents and Media
3.2.2. Bacterial Strains
3.2.3. MIC Determination
3.2.4. Real-Time Ethidium Bromide Accumulation Assay
3.2.5. Measuring Biofilm Formation Using Crystal Violet
3.2.6. Antibiotic Potentiation Assay
3.2.7. Cytotoxicity Assay
3.2.8. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report 2022; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Podolsky, S.H. The evolving response to antibiotic resistance (1945–2018). Palgrave Commun. 2018, 4, 124. [Google Scholar] [CrossRef]
- Domínguez, D.C.; Meza-Rodriguez, S.M. 16-Development of antimicrobial resistance: Future challenges. In Pharmaceuticals and Personal Care Products: Waste Management and Treatment Technology; Prasad, M.N.V., Vithanage, M., Kapley, A., Eds.; Butterworth-Heinemann: Oxford, UK, 2019; pp. 383–408. [Google Scholar]
- OECD. Stemming the Superbug Tide: Just A Few Dollars More. In OECD Health Policy Studies; OECD Publishing: Paris, France, 2018. [Google Scholar]
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Michael, C.A.; Dominey-Howes, D.; Labbate, M. The Antimicrobial Resistance Crisis: Causes, Consequences, and Management. Front. Public Health 2014, 2, 145. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T. Who will develop new antibacterial agents? Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130430. [Google Scholar] [CrossRef]
- Christaki, E.; Marcou, M.; Tofarides, A. Antimicrobial Resistance in Bacteria: Mechanisms, Evolution, and Persistence. J. Mol. Evol. 2020, 88, 26–40. [Google Scholar] [CrossRef]
- Khameneh, B.; Diab, R.; Ghazvini, K.; Fazly Bazzaz, B.S. Breakthroughs in bacterial resistance mechanisms and the potential ways to combat them. Microb. Pathog. 2016, 95, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Deng, Z.; Yan, A. Bacterial multidrug efflux pumps: Mechanisms, physiology and pharmacological exploitations. Biochem. Biophys. Res. Commun. 2014, 453, 254–267. [Google Scholar] [CrossRef]
- Kvist, M.; Hancock, V.; Klemm, P. Inactivation of Efflux Pumps Abolishes Bacterial Biofilm Formation. Appl. Environ. Microbiol. 2008, 74, 7376–7382. [Google Scholar] [CrossRef]
- Alav, I.; Sutton, J.M.; Rahman, K.M. Role of bacterial efflux pumps in biofilm formation. J. Antimicrob. Chemother. 2018, 73, 2003–2020. [Google Scholar] [CrossRef]
- Du, D.; Wang, Z.; James, N.R.; Voss, J.E.; Klimont, E.; Ohene-Agyei, T.; Venter, H.; Chiu, W.; Luisi, B.F. Structure of the AcrAB–TolC multidrug efflux pump. Nature 2014, 509, 512–515. [Google Scholar] [CrossRef]
- Tikhonova, E.B.; Zgurskaya, H.I. AcrA, AcrB, and TolC of Escherichia coli Form a Stable Intermembrane Multidrug Efflux Complex . J. Biol. Chem. 2004, 279, 32116–32124. [Google Scholar] [CrossRef] [PubMed]
- Chetri, S.; Bhowmik, D.; Paul, D.; Pandey, P.; Chanda, D.D.; Chakravarty, A.; Bora, D.; Bhattacharjee, A. AcrAB-TolC efflux pump system plays a role in carbapenem non-susceptibility in Escherichia coli. BMC Microbiol. 2019, 19, 210. [Google Scholar] [CrossRef] [PubMed]
- Chollet, R.; Chevalier, J.; Bryskier, A.; Pagès, J.-M. The AcrAB-TolC Pump Is Involved in Macrolide Resistance but not in Telithromycin Efflux in Enterobacter aerogenes and Escherichia coli. Antimicrob. Agents Chemother. 2004, 48, 3621–3624. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Wang, H.; Zou, L.; Zhang, A.; Yang, X.; Guan, Z. Evaluation and Target Validation of Indole Derivatives as Inhibitors of the AcrAB-TolC Efflux Pump. Biosci. Biotechnol. Biochem. 2010, 74, 2237–2241. [Google Scholar] [CrossRef] [PubMed]
- Durand, G.A.; Raoult, D.; Dubourg, G. Antibiotic discovery: History, methods and perspectives. Int. J. Antimicrob. Agents 2019, 53, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Resende, D.I.S.P.; Boonpothong, P.; Sousa, E.; Kijjoa, A.; Pinto, M.M.M. Chemistry of the fumiquinazolines and structurally related alkaloids. Nat. Prod. Rep. 2019, 36, 7–34. [Google Scholar] [CrossRef]
- Long, S.; Duarte, D.; Carvalho, C.; Oliveira, R.; Santarém, N.; Palmeira, A.; Resende, D.I.S.P.; Silva, A.M.S.; Moreira, R.; Kijjoa, A.; et al. Indole-Containing Pyrazino[2,1-b]quinazoline-3,6-diones Active against Plasmodium and Trypanosomatids. ACS Med. Chem. Lett. 2022, 13, 225–235. [Google Scholar] [CrossRef]
- Long, S.; Resende, D.I.S.P.; Kijjoa, A.; Silva, A.M.S.; Fernandes, R.; Xavier, C.P.R.; Vasconcelos, M.H.; Sousa, E.; Pinto, M.M.M. Synthesis of New Proteomimetic Quinazolinone Alkaloids and Evaluation of Their Neuroprotective and Antitumor Effects. Molecules 2019, 24, 534. [Google Scholar] [CrossRef]
- Barreiro, S.; Silva, B.; Long, S.; Pinto, M.; Remião, F.; Sousa, E.; Silva, R. Fiscalin Derivatives as Potential Neuroprotective Agents. Pharmaceutics 2022, 14, 1456. [Google Scholar] [CrossRef]
- Peng, J.; Lin, T.; Wang, W.; Xin, Z.; Zhu, T.; Gu, Q.; Li, D. Antiviral Alkaloids Produced by the Mangrove-Derived Fungus Cladosporium sp. PJX-41. J. Nat. Prod. 2013, 76, 1133–1140. [Google Scholar] [CrossRef]
- Li, X.-J.; Zhang, Q.; Zhang, A.-L.; Gao, J.-M. Metabolites from Aspergillus fumigatus, an Endophytic Fungus Associated with Melia azedarach, and Their Antifungal, Antifeedant, and Toxic Activities. J. Agric. Food Chem. 2012, 60, 3424–3431. [Google Scholar] [CrossRef] [PubMed]
- Bessa, L.J.; Buttachon, S.; Dethoup, T.; Martins, R.; Vasconcelos, V.; Kijjoa, A.; Martins da Costa, P. Neofiscalin A and fiscalin C are potential novel indole alkaloid alternatives for the treatment of multidrug-resistant Gram-positive bacterial infections. FEMS Microbiol. Lett. 2016, 363, fnw150. [Google Scholar] [CrossRef] [PubMed]
- Long, S.; Resende, D.I.S.P.; Palmeira, A.; Kijjoa, A.; Silva, A.M.S.; Tiritan, M.E.; Pereira-Terra, P.; Freitas-Silva, J.; Barreiro, S.; Silva, R.; et al. New marine-derived indolymethyl pyrazinoquinazoline alkaloids with promising antimicrobial profiles. RSC Adv. 2020, 10, 31187–31204. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ganesan, A. Total Synthesis of the Fumiquinazoline Alkaloids: Solution-Phase Studies1. J. Org. Chem. 2000, 65, 1022–1030. [Google Scholar] [CrossRef]
- Gamon, L.F.; White, J.M.; Wille, U. Oxidative damage of aromatic dipeptides by the environmental oxidants NO2 and O3. Org. Biomol. Chem. 2014, 12, 8280–8287. [Google Scholar] [CrossRef]
- Long, S.; Resende, D.; Kijjoa, A.; Silva, A.M.S.; Pina, A.; Fernández-Marcelo, T.; Vasconcelos, M.H.; Sousa, E.; Pinto, M.M.M. Antitumor Activity of Quinazolinone Alkaloids Inspired by Marine Natural Products. Mar. Drugs 2018, 16, 261. [Google Scholar] [CrossRef]
- Kantharaju; Patil, B.S.; Suresh Babu, V.V. Synthesis of Fmoc-amino acid chlorides assisted by ultrasonication, a rapid approach. Lett. Pept. Sci. 2002, 9, 227–229. [Google Scholar] [CrossRef]
- Kshirsagar, U.A.; Mhaske, S.B.; Argade, N.P. Hexamethyldisilazane-iodine induced intramolecular dehydrative cyclization of diamides: A general access to natural and unnatural quinazolinones. Tetrahedron Lett. 2007, 48, 3243–3246. [Google Scholar] [CrossRef]
- He, F.; Snider, B.B. Rearrangement of 4-Imino-4H-3,1-benzoxazines to 4-Quinazolinones via Amidine Carboxamides. J. Org. Chem. 1999, 64, 1397–1399. [Google Scholar] [CrossRef]
- Hernández, F.; Buenadicha, F.L.; Avendaño, C.; Söllhuber, M. 1-Alkyl-2,4-dihydro-1H-pyrazino[2,1-b]quinazoline-3,6-diones as glycine templates. Synthesis of Fiscalin B. Tetrahedron Asymmetry 2002, 12, 3387–3398. [Google Scholar] [CrossRef]
- Durães, F.; Palmeira, A.; Cruz, B.; Freitas-Silva, J.; Szemerédi, N.; Gales, L.; da Costa, P.M.; Remião, F.; Silva, R.; Pinto, M.; et al. Antimicrobial Activity of a Library of Thioxanthones and Their Potential as Efflux Pump Inhibitors. Pharmaceuticals 2021, 14, 572. [Google Scholar] [CrossRef] [PubMed]
- Resende, D.I.S.P.; Pereira-Terra, P.; Moreira, J.; Freitas-Silva, J.; Lemos, A.; Gales, L.; Pinto, E.; de Sousa, M.E.; da Costa, P.M.; Pinto, M.M.M. Synthesis of a Small Library of Nature-Inspired Xanthones and Study of Their Antimicrobial Activity. Molecules 2020, 25, 2405. [Google Scholar] [CrossRef] [PubMed]
- Viveiros, M.; Martins, A.; Paixão, L.; Rodrigues, L.; Martins, M.; Couto, I.; Fähnrich, E.; Kern, W.V.; Amaral, L. Demonstration of intrinsic efflux activity of Escherichia coli K-12 AG100 by an automated ethidium bromide method. Int. J. Antimicrob. Agents 2008, 31, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Paixão, L.; Rodrigues, L.; Couto, I.; Martins, M.; Fernandes, P.; de Carvalho, C.C.C.R.; Monteiro, G.A.; Sansonetty, F.; Amaral, L.; Viveiros, M. Fluorometric determination of ethidium bromide efflux kinetics in Escherichia coli. J. Biol. Eng. 2009, 3, 18. [Google Scholar] [CrossRef]
- Annamária, K.; Ágnes Míra, S.; Ryosuke, S.; Genki, W.; Masami, K.; Joseph, M.; Gabriella, S. Fluorinated Beta-diketo Phosphorus Ylides Are Novel Efflux Pump Inhibitors in Bacteria. Vivo 2016, 30, 813. [Google Scholar]
- CLSI. Performance Standards for Antimicrobial Disk Susceptibility Tests, 11th ed.; CLSI: Wayne, PA, USA, 2012. [Google Scholar]
- Shi, X.; Chen, M.; Yu, Z.; Bell, J.M.; Wang, H.; Forrester, I.; Villarreal, H.; Jakana, J.; Du, D.; Luisi, B.F.; et al. In situ structure and assembly of the multidrug efflux pump AcrAB-TolC. Nat. Commun. 2019, 10, 2635. [Google Scholar] [CrossRef]
- Yan, N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 2013, 38, 151–159. [Google Scholar] [CrossRef]
- Aron, Z.; Opperman, T.J. The hydrophobic trap—The Achilles heel of RND efflux pumps. Res. Microbiol. 2018, 169, 393–400. [Google Scholar] [CrossRef]
- Zárate, S.G.; Morales, P.; Świderek, K.; Bolanos-Garcia, V.M.; Bastida, A. A Molecular Modeling Approach to Identify Novel Inhibitors of the Major Facilitator Superfamily of Efflux Pump Transporters. Antibiotics 2019, 8, 25. [Google Scholar] [CrossRef]
- Durães, F.; Resende, D.I.S.P.; Palmeira, A.; Szemerédi, N.; Pinto, M.M.M.; Spengler, G.; Sousa, E. Xanthones Active against Multidrug Resistance and Virulence Mechanisms of Bacteria. Antibiotics 2021, 10, 600. [Google Scholar] [CrossRef]
- Simões, R.R.; Aires-de-Sousa, M.; Conceição, T.; Antunes, F.; da Costa, P.M.; de Lencastre, H. High Prevalence of EMRSA-15 in Portuguese Public Buses: A Worrisome Finding. PLoS ONE 2011, 6, e17630. [Google Scholar] [CrossRef] [PubMed]
- Bessa, L.J.; Barbosa-Vasconcelos, A.; Mendes, Â.; Vaz-Pires, P.; Martins da Costa, P. High prevalence of multidrug-resistant Escherichia coli and Enterococcus spp. in river water, upstream and downstream of a wastewater treatment plant. J. Water Health 2014, 12, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Martins da Costa, P.; Vaz-Pires, P.; Bernardo, F. Antimicrobial resistance in Enterococcus spp. isolated in inflow, effluent and sludge from municipal sewage water treatment plants. Water Res. 2006, 40, 1735–1740. [Google Scholar] [CrossRef]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. In Approved Standard, 9th ed.; CLSI: Wayne, PA, USA, 2012. [Google Scholar]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Matsumoto, T.; Yamaguchi, A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 2006, 443, 173–179. [Google Scholar] [CrossRef]
- Mikolosko, J.; Bobyk, K.; Zgurskaya, H.I.; Ghosh, P. Conformational flexibility in the multidrug efflux system protein AcrA. Structure 2006, 14, 577–587. [Google Scholar] [CrossRef]
- Koronakis, V.; Sharff, A.; Koronakis, E.; Luisi, B.; Hughes, C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 2000, 405, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of Three-Dimensional Structural Information of Biological Macromolecules. Acta Crystallogr. Sect. D 1998, 54, 1078–1084. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Shaheen, A.; Afridi, W.A.; Mahboob, S.; Sana, M.; Zeeshan, N.; Ismat, F.; Mirza, O.; Iqbal, M.; Rahman, M. Reserpine Is the New Addition into the Repertoire of AcrB Efflux Pump Inhibitors. Mol. Biol. 2019, 53, 596–605. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169.
- Zimmermann, S.; Klinger-Strobel, M.; Bohnert, J.A.; Wendler, S.; Rödel, J.; Pletz, M.W.; Löffler, B.; Tuchscherr, L. Clinically Approved Drugs Inhibit the Staphylococcus aureus Multidrug NorA Efflux Pump and Reduce Biofilm Formation. Front. Microbiol. 2019, 10, 2762. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
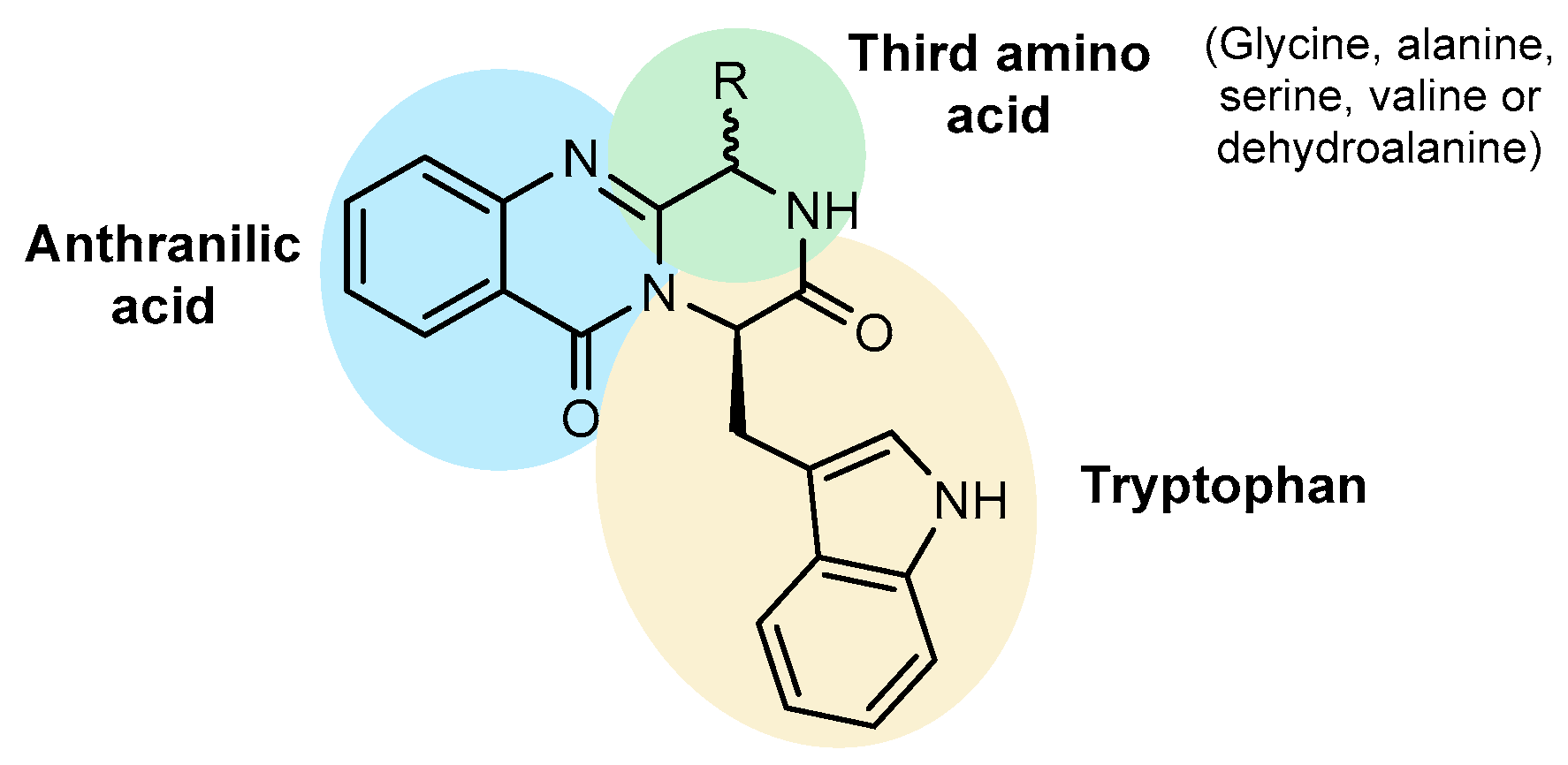{kind=link}
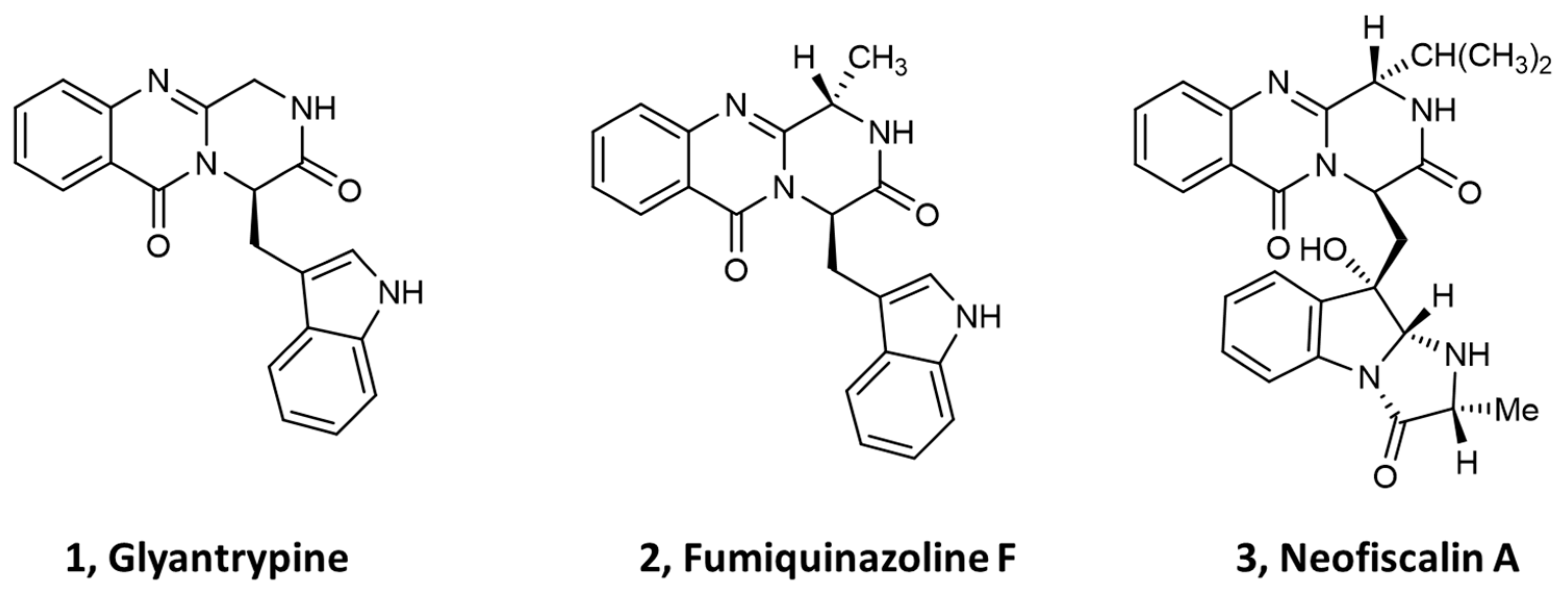{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Peak Purity | Chromatographic Purity | [αD25] | Enantiomeric Excess (ee) |
|---|---|---|---|---|
| 1 | 97.8% | 100% | −390 (c 0.033, THF) | >99% |
| 2 | 95.3% | 100% | −334 (c 0.030, THF) | >99% |
| 12a | 99.9% | 97.9% | −464 (c 0.037, THF) | >99% |
| 12b | 99.9% | 94.3% | −273 (c 0.037, THF) | >99% |
| 12c | 94.2% | 100% | +450 (c 0.04, THF) | >99% |
| 12d | 97.4% | 100% | +510 (c 0.033, THF) | >99% |
| S. aureus Strains | E. faecalis Strains | ||||||
|---|---|---|---|---|---|---|---|
| Compound | A | B | C | D | E | F | G |
| 1 | >100 | >100 | >100 | >100 | >100 | n.t | n.t |
| 2 | >100 | >100 | >100 | >100 | >100 | n.t | n.t |
| 12a | >100 | 77 (32) | >100 | >100 | 77 (32) | >100 | >100 |
| 12b | 77 (32) | 77 (32) | 100 (41.8) | >100 | 38 (16) | >100 | >100 |
| 12c | 33 (16) | 33(16) | 12.50 (6.1) | 33–66 (16–32) | 33 (16) | >100 | >100 |
| 12d | 32 (16) | 16 (8) | 25 (12.6) | 64 (32) | 64 (32) | >100 | >100 |
| 4 | n.t | n.t | >100 | n.t | n.t | n.t | n.t |
| 13a | n.t | n.t | >100 | n.t | n.t | n.t | n.t |
| 13b | n.t | n.t | >100 | n.t | n.t | n.t | n.t |
| 13c | n.t | n.t | 100 (40.05) | n.t | n.t | n.t | n.t |
| 13d | n.t | n.t | >100 | n.t | n.t | n.t | n.t |
| E. coli AG100 | E. coli AG100A | S. aureus 272123 | ||||
|---|---|---|---|---|---|---|
| Compound | Concentration (μM) | RFI ± SD | Concentration (μM) | RFI ± SD | Concentration (μM) | RFI ± SD |
| 1 | 100 50 | −0.04 ± 0.10 −0.24 ± 0.05 | 100 | 0.12 ± 0.01 | 100 | −0.01 ± 0.04 |
| 50 | 0.23 ± 0.02 | |||||
| 2 | 100 50 | 0.17 ± 0.10 −0.31 ± 0.02 | 100 | 0.26 ± 0.10 | 100 | 0.05 ±0.02 |
| 50 | −0.01 ± 0.02 | |||||
| 12a | 100 50 | 2.45 ± 0.26 −0.17 ± 0.02 | 100 | 0.59 ± 0.03 | 100 | 1.03 ± 0.02 |
| 50 | 0.63 ± 0.04 | |||||
| 12b | 100 50 | 1.47 ± 0.12 −0.07 ± 0.06 | 100 | 0.77 ± 0.17 | 50 | 0.96 ± 0.07 |
| 12c | 100 | 3.60 ± 0.01 | 100 | 4.40 ± 1.78 | 6.25 | 0.49 ± 0.01 |
| 50 | 0.24 ± 0.16 | |||||
| 12d | 100 | 0.32 ± 0.10 | 100 | 0.23 ± 0.05 | 12.5 | 0.34 ± 0.01 |
| 4 | 50 | 0.01 ± 0.02 | 50 | 0.26 ± 0.07 | 50 | 1.13 ± 0.20 |
| 13a | 50 | 0.38 ± 0.01 | 50 | 0.48 ± 0.58 | 50 | 2.39 ± 0.09 |
| 13b | 50 | 0.52 ± 0.18 | 50 | 0.12 ± 0.05 | 50 | 1.53 ± 0.02 |
| 13c | 50 | 0.17 ± 0.02 | 50 | 0.13 ± 0.05 | 50 | 0.94 ± 0.04 |
| 13d | 50 | −0.01 ± 0.03 | 50 | 0.55 ± 0.29 | 50 | 0.19 ± 0.08 |
| CCCP | 50 | 1.56 ± 0.01 | 50 | 1.13 ± 0.02 | - | - |
| RES | - | - | - | - | 25 | 0.68 ± 0.05 |
| Cytotoxicity on NIH/3T3 | |
|---|---|
| Compounds | IC50 ± SD (µM) |
| 1 | >100 |
| 2 | >100 |
| 12a | 52.20 ± 0.75 |
| 12b | 58.04 ± 1.58 |
| 12c | 46.15 ± 0.66 |
| 12d | 47.23 ± 1.55 |
| DOXO | 3.72 ± 0.15 |
| DMSO | >2% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, M.C.; Szemerédi, N.; Durães, F.; Long, S.; Resende, D.I.S.P.; Martins da Costa, P.; Pinto, M.; Spengler, G.; Sousa, E. Effect of Indole-Containing Pyrazino[2,1-b]quinazoline-3,6-diones in the Virulence of Resistant Bacteria. Antibiotics 2023, 12, 922. https://doi.org/10.3390/antibiotics12050922
Almeida MC, Szemerédi N, Durães F, Long S, Resende DISP, Martins da Costa P, Pinto M, Spengler G, Sousa E. Effect of Indole-Containing Pyrazino[2,1-b]quinazoline-3,6-diones in the Virulence of Resistant Bacteria. Antibiotics. 2023; 12(5):922. https://doi.org/10.3390/antibiotics12050922
Chicago/Turabian StyleAlmeida, Mariana C., Nikoletta Szemerédi, Fernando Durães, Solida Long, Diana I. S. P. Resende, Paulo Martins da Costa, Madalena Pinto, Gabriella Spengler, and Emília Sousa. 2023. "Effect of Indole-Containing Pyrazino[2,1-b]quinazoline-3,6-diones in the Virulence of Resistant Bacteria" Antibiotics 12, no. 5: 922. https://doi.org/10.3390/antibiotics12050922