
Neutralizing Carbapenem Resistance by Co-Administering Meropenem with Novel β-Lactam-Metallo-β-Lactamase Inhibitors
 , , , , , , , ,
, , , , , , , ,  , and
, and
Abstract
:1. Introduction
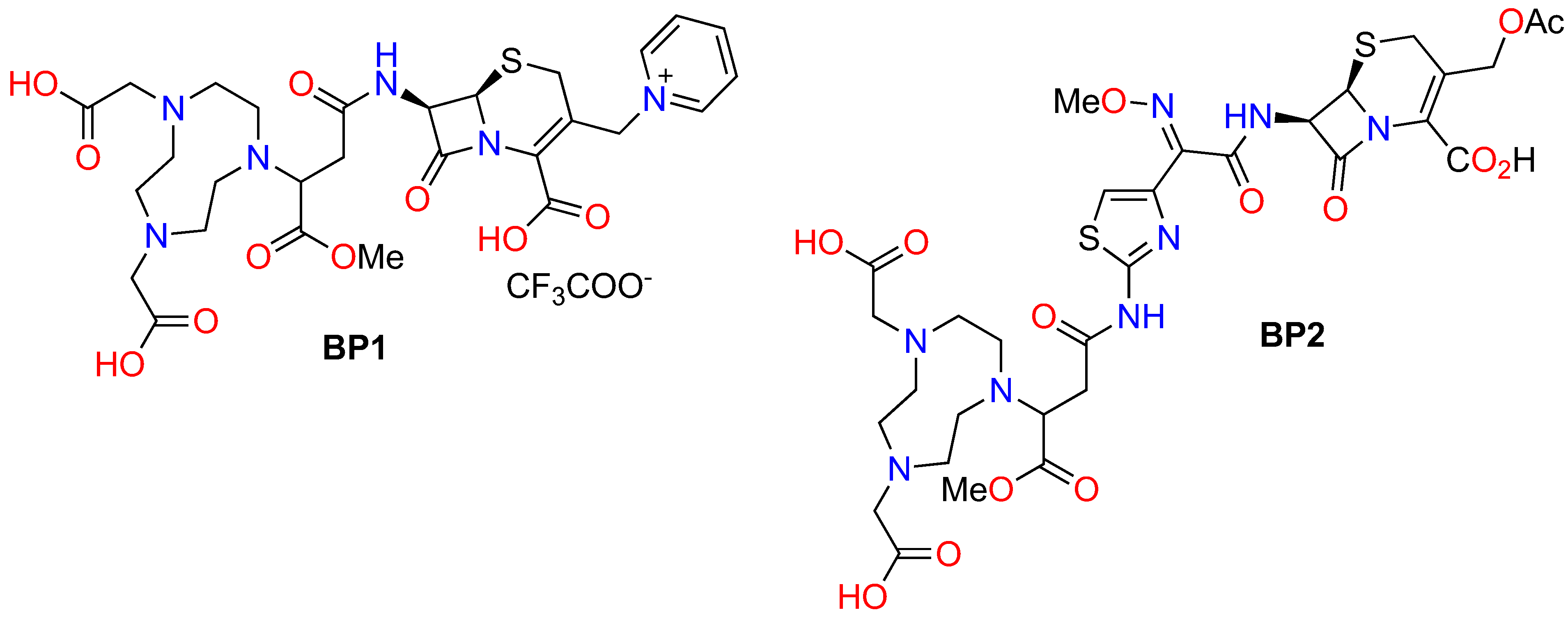
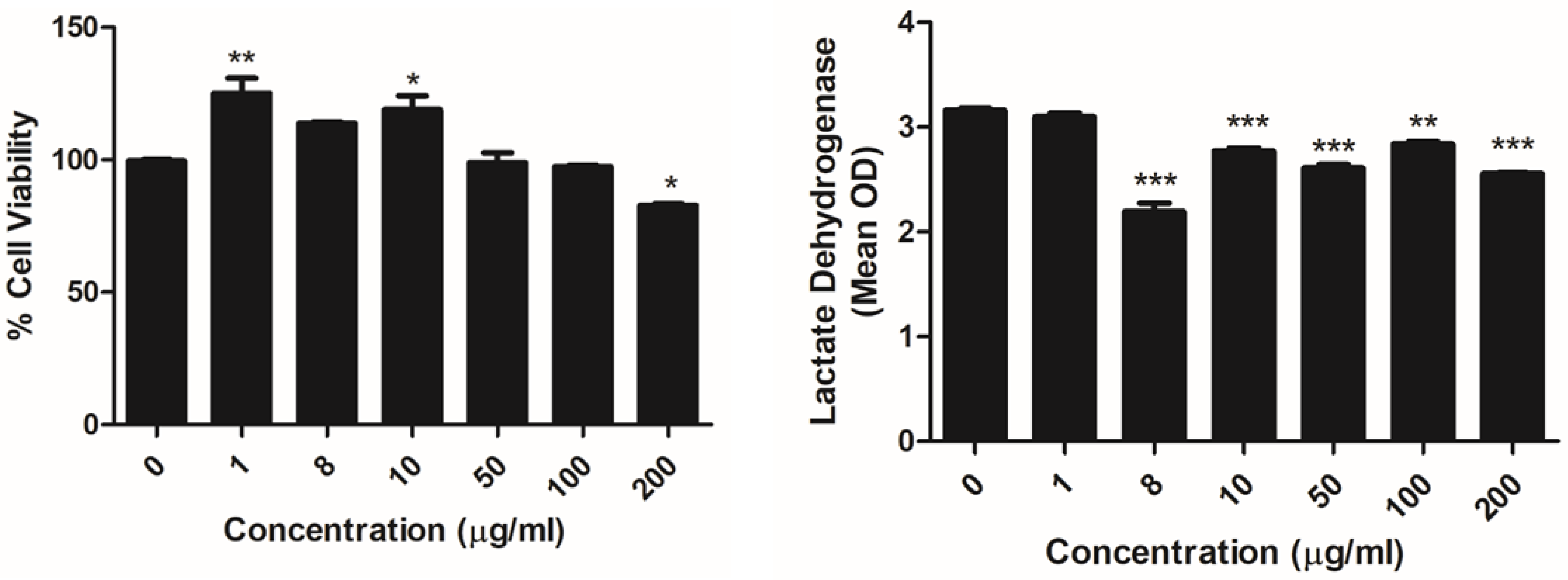
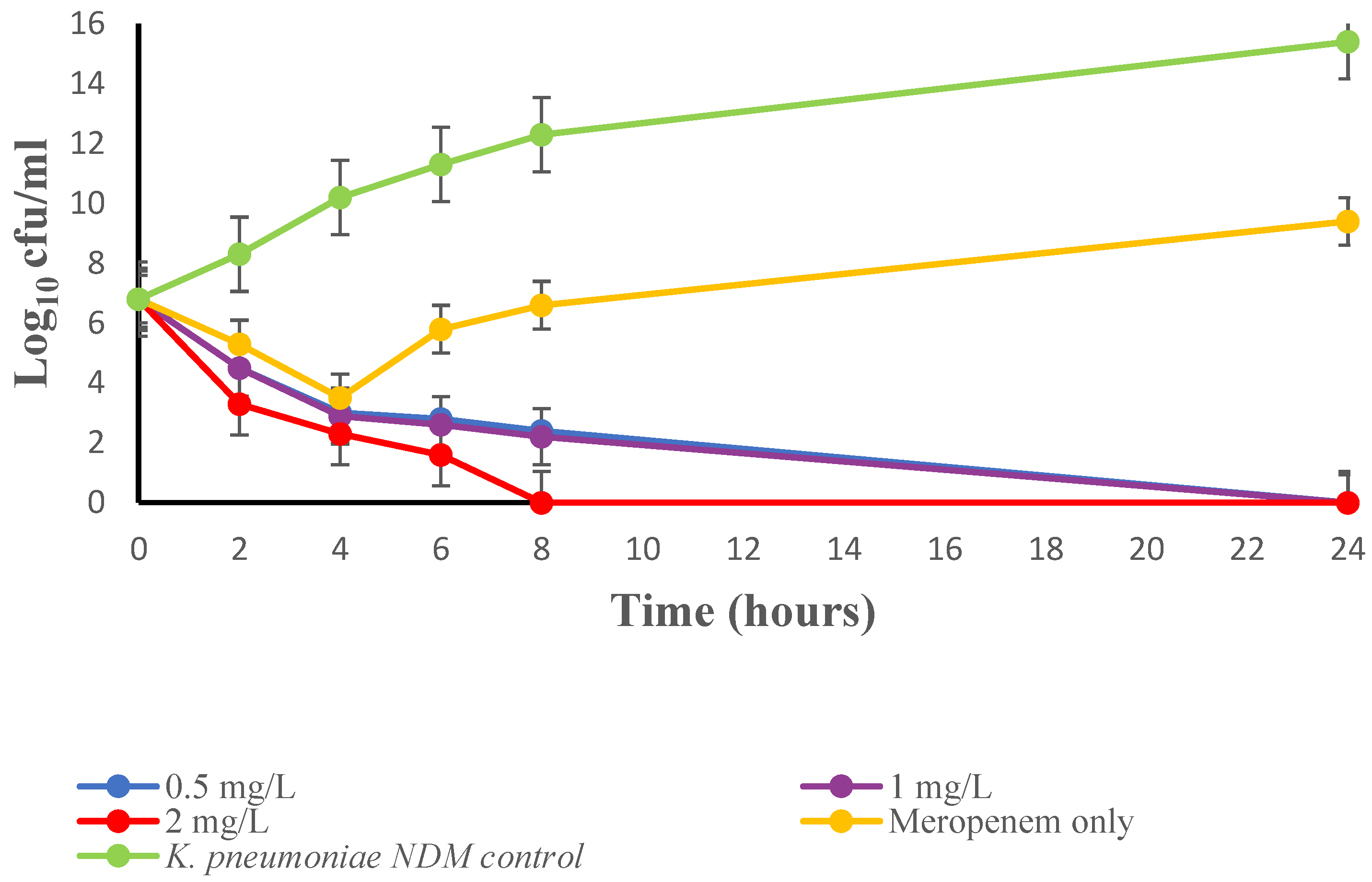
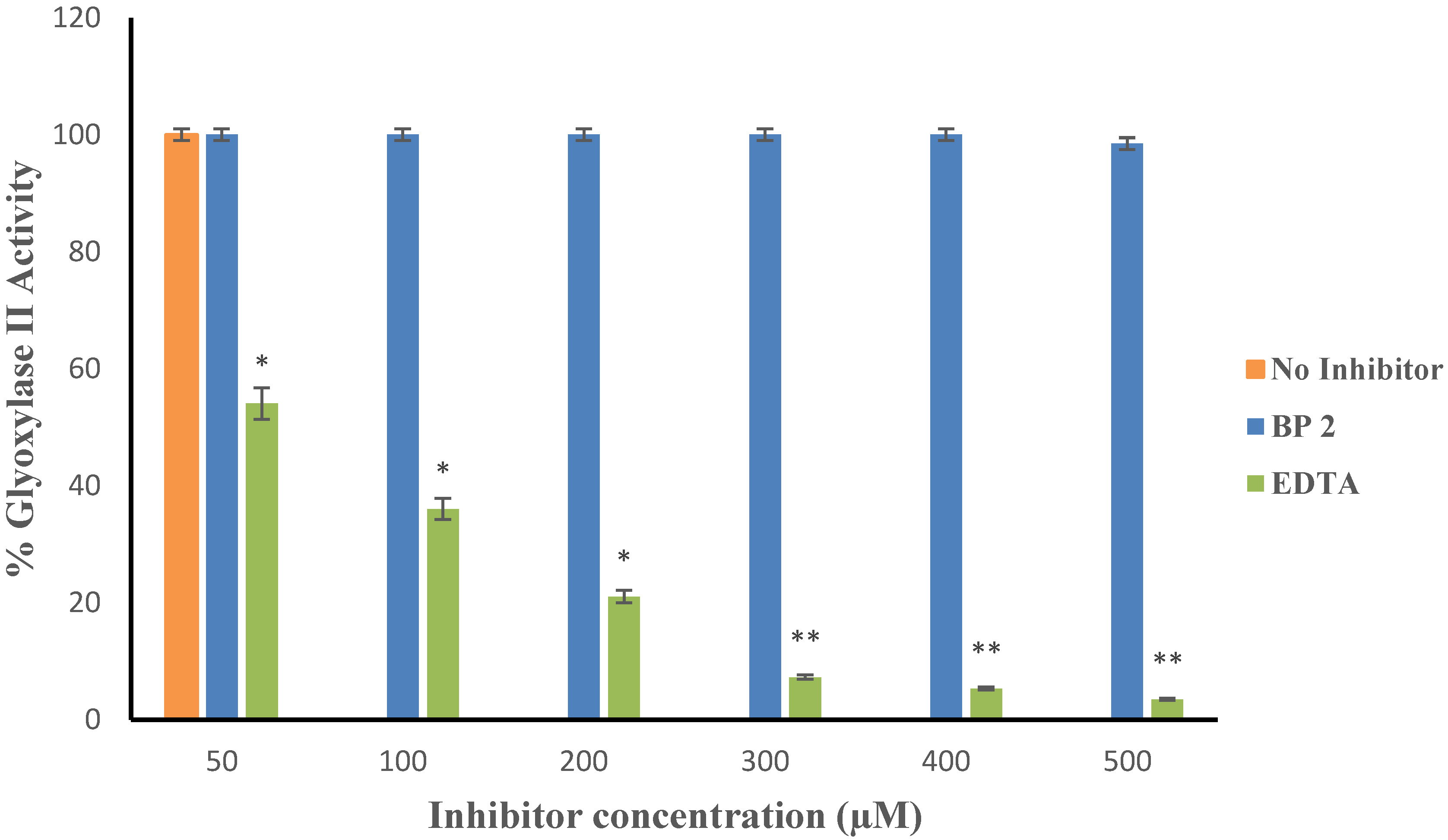
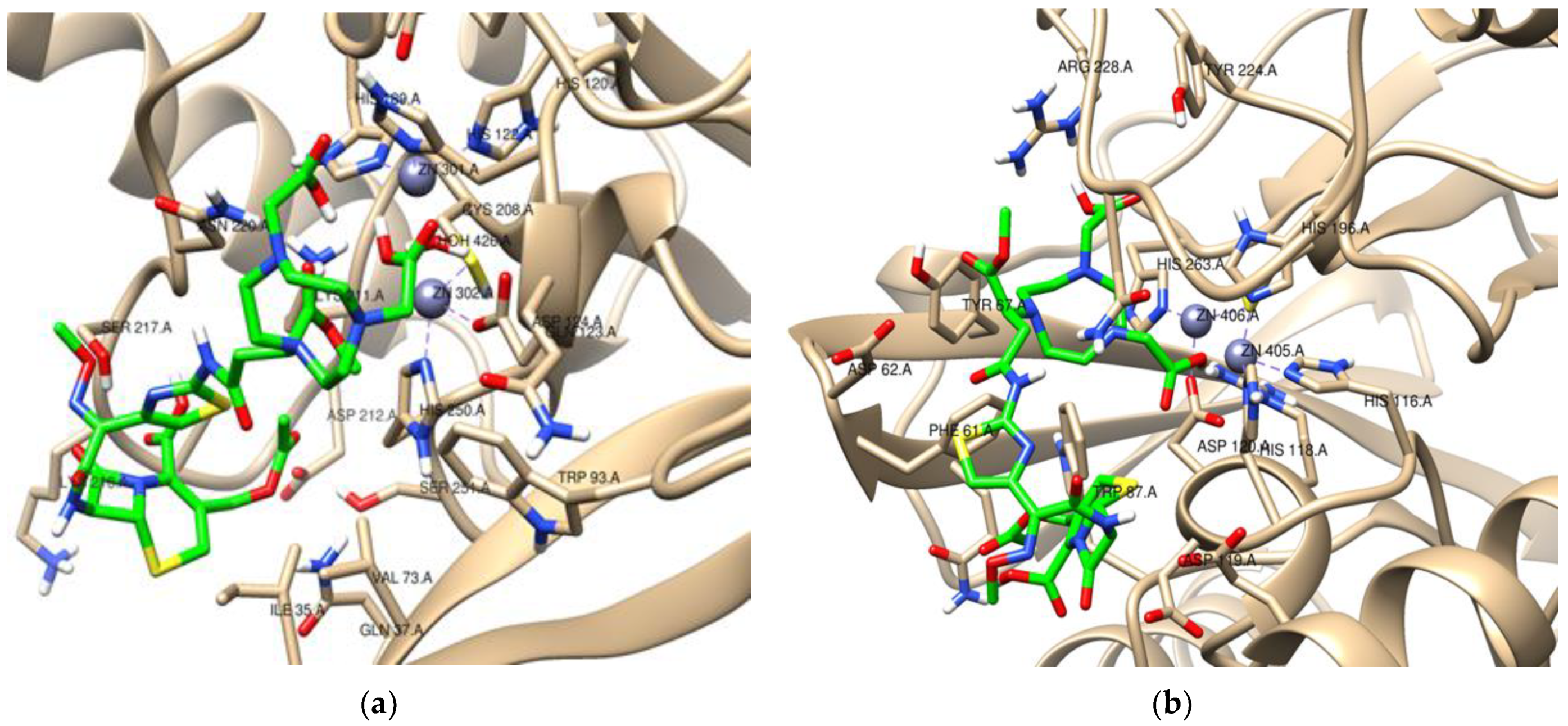
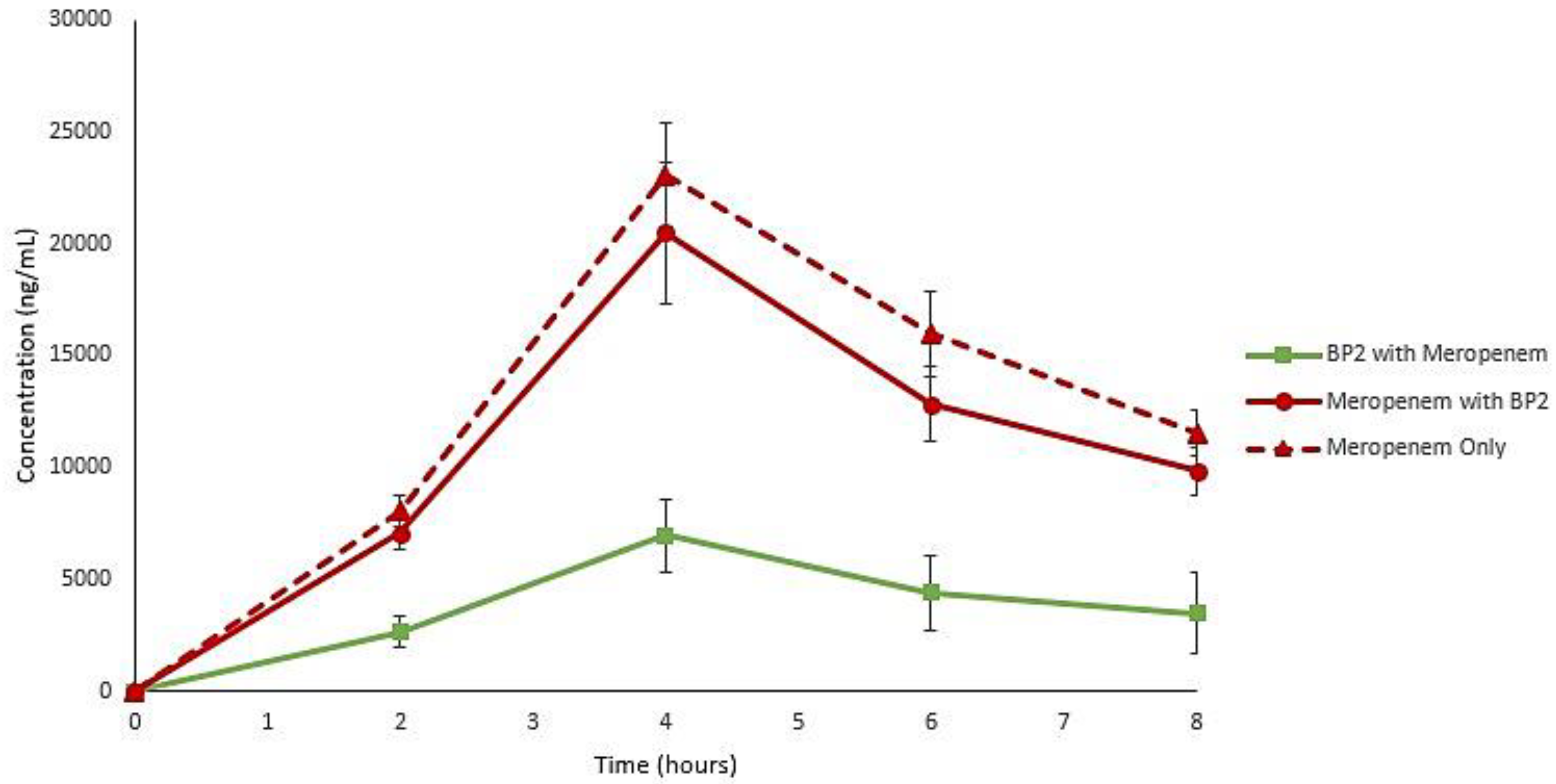
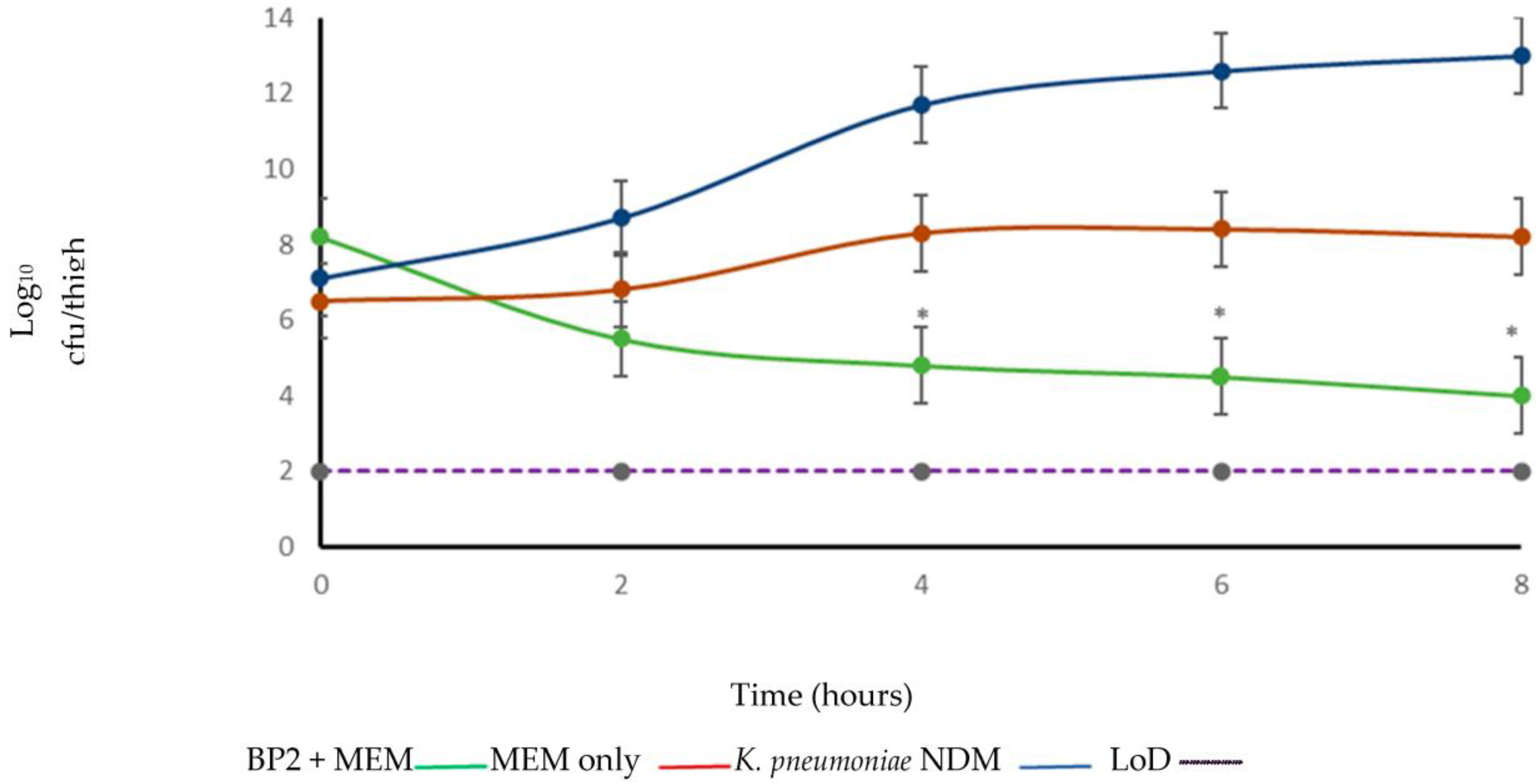
2. Results
3. Discussion
4. Materials and Methods
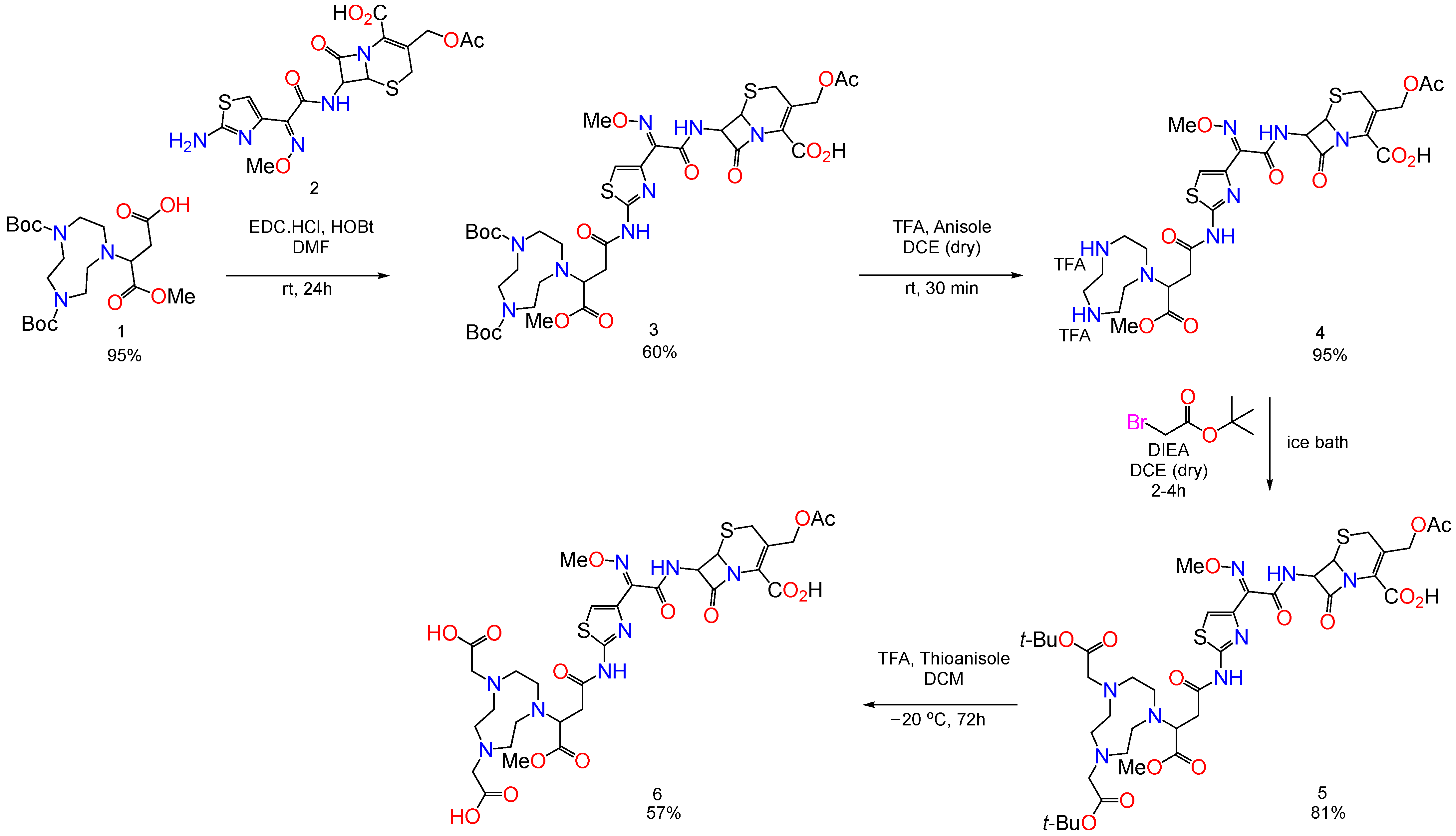
4.1. Synthesis of MBLIs
Synthesis of BP2
4.2. Bacterial Source
4.3. Drug Susceptibility Testing
4.3.1. Broth Microdilution Assay
4.3.2. Checkerboard Assay
4.3.3. Effects of Human Serum
4.3.4. Time-Kill Study
4.4. Cytotoxicity Assay
4.4.1. Cell Culture
4.4.2. MTT Assay
4.4.3. LDH Assay
4.5. Enzyme Assays
4.5.1. Inhibition of Kinetics
4.5.2. Non-Specific Binding of the Inhibitor to Zinc in Non-MBLs
4.6. Ethical Statement
4.7. In Vivo Efficacy Study
4.8. LC-MS Quantification
4.9. Computational Studies
4.10. Statistical Analyses
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mojica, M.F.; Rossi, M.-A.; Vila, A.J.; Bonomo, R.A. The urgent need for metallo-β-lactamase inhibitors: An unattended global threat. Lancet Infect. Dis. 2022, 22, e28–e34. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.-S.; Gould, I.M.; Lee, W.-S.; Hsueh, P.-R. New drugs for multidrug-resistant gram-negative organisms: Time for stewardship. Drugs 2019, 79, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Song, X.; Li, M.; Yu, Z.; Cheng, W.; Yu, Z.; Zhang, W.; Zhang, Y.; Shen, A.; Sun, H. Global Spread of Carbapenem-Resistant Enterobacteriaceae: Epidemiological Features, Resistance Mechanisms, Detection and Therapy. Microbiol. Res. 2022, 266, 127249. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Bonomo, R.A. Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef] [Green Version]
- Toussaint, K.A.; Gallagher, J.C. β-Lactam/β-lactamase inhibitor combinations: From then to now. Ann. Pharm. 2015, 49, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Logan, L.K.; Weinstein, R.A. The epidemiology of carbapenem-resistant Enterobacteriaceae: The impact and evolution of a global menace. J. Infect. Dis. 2017, 215, S28–S36. [Google Scholar] [CrossRef] [Green Version]
- Bush, K.; Jacoby, G.A. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Tsivkovski, R.; Totrov, M.; Lomovskaya, O. Biochemical characterization of QPX7728, a new ultrabroad-spectrum beta-lactamase inhibitor of serine and metallo-beta-lactamases. Antimicrob. Agents Chemother. 2020, 64, e00130-20. [Google Scholar] [CrossRef] [Green Version]
- Cornaglia, G.; Giamarellou, H.; Rossolini, G.M. Metallo-β-lactamases: A last frontier for β-lactams? Lancet Infect. Dis. 2011, 11, 381–393. [Google Scholar] [CrossRef]
- Walsh, T.R.; Toleman, M.A.; Poirel, L.; Nordmann, P. Metallo-β-lactamases: The quiet before the storm? Clin. Microbiol. Rev. 2005, 18, 306–325. [Google Scholar] [CrossRef] [Green Version]
- Carcione, D.; Siracusa, C.; Sulejmani, A.; Leoni, V.; Intra, J. Old and new beta-lactamase inhibitors: Molecular structure, mechanism of action, and clinical Use. Antibiotics 2021, 10, 995. [Google Scholar] [CrossRef] [PubMed]
- King, D.T.; Strynadka, N.C. Targeting metallo-β-lactamase enzymes in antibiotic resistance. Future Med. Chem. 2013, 5, 1243–1263. [Google Scholar] [CrossRef] [PubMed]
- Shakil, S.; Azhar, E.; Tabrez, S.; Kamal, M.; Jabir, N.; Abuzenadah, A.; Damanhouri, G.; Alam, Q. New Delhi metallo-β-lactamase (NDM-1): An updates. J. Chemother. 2011, 23, 263–265. [Google Scholar] [CrossRef]
- Johnson, A.P.; Woodford, N. Global spread of antibiotic resistance: The example of New Delhi metallo-β-lactamase (NDM)-mediated carbapenem resistance. J. Med. Microbiol. 2013, 62, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-F.; Chou, K.-C. Metallo-β-lactamases: Structural features, antibiotic recognition, inhibition, and inhibitor design. Curr. Top. Med. Chem. 2013, 13, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Hunter, P.A.; Coleman, K.; Fisher, J.; Taylor, D. In vitro synergistic properties of clavulanic acid, with ampicillin, amoxycillin and ticarcillin. J. Antimicrob. Chemother. 1980, 6, 455–470. [Google Scholar] [CrossRef]
- De Koning, G.; Tio, D.; Coster, J.; Coutinho, R.; Ansink-Schipper, M. The combination of clavulanic acid and amoxycillin (Augmentin) in the treatment of patients infected with penicillinase producing gonococci. J. Antimicrob. Chemother. 1981, 8, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Temkin, E.; Torre-Cisneros, J.; Beovic, B.; Benito, N.; Giannella, M.; Gilarranz, R.; Jeremiah, C.; Loeches, B.; Machuca, I.; Jiménez-Martín, M.J. Ceftazidime-avibactam as salvage therapy for infections caused by carbapenem-resistant organisms. Antimicrob. Agents Chemother. 2017, 61, e01964-16. [Google Scholar] [CrossRef] [Green Version]
- Hackel, M.A.; Lomovskaya, O.; Dudley, M.N.; Karlowsky, J.A.; Sahm, D.F. In vitro activity of meropenem-vaborbactam against clinical isolates of KPC-positive Enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62, e01904-17. [Google Scholar] [CrossRef] [Green Version]
- Meletiadis, J.; Paranos, P.; Georgiou, P.-C.; Vourli, S.; Antonopoulou, S.; Michelaki, A.; Vagiakou, E.; Pournaras, S. In vitro comparative activity of the new beta-lactamase inhibitor taniborbactam with cefepime or meropenem against Klebsiella pneumoniae and cefepime against Pseudomonas aeruginosa metallo-beta-lactamase-producing clinical isolates. Int. J. Antimicrob. Agents 2021, 58, 106440. [Google Scholar] [CrossRef]
- Reddy, N.; Shungube, M.; Arvidsson, P.I.; Baijnath, S.; Kruger, H.G.; Govender, T.; Naicker, T. A 2018–2019 patent review of metallo beta-lactamase inhibitors. Expert Opin. Ther. Pat. 2020, 30, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Mojica, M.F.; Bonomo, R.A.; Fast, W. B1-metallo-β-lactamases: Where do we stand? Curr. Drug Targets 2016, 17, 1029–1050. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Rubio-Aparicio, D.; Nelson, K.; Sun, D.; Tsivkovski, R.; Castanheira, M.; Lindley, J.; Loutit, J.; Dudley, M. In vitro activity of the ultrabroad-spectrum beta-lactamase inhibitor QPX7728 in combination with multiple beta-lactam antibiotics against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2021, 65, e00210-21. [Google Scholar] [CrossRef] [PubMed]
- Buynak, J.D. β-Lactamase inhibitors: A review of the patent literature (2010–2013). Expert Opin. Ther. Pat. 2013, 23, 1469–1481. [Google Scholar] [CrossRef]
- Talbot, G.H.; Jezek, A.; Murray, B.E.; Jones, R.N.; Ebright, R.H.; Nau, G.J.; Rodvold, K.A.; Newland, J.G.; Boucher, H.W.; The Infectious Diseases Society of America. The Infectious Diseases Society of America’s 10 × ’20 initiative (10 new systemic antibacterial agents US Food and Drug Administration approved by 2020): Is 20 × ’20 a possibility? Clin. Infect. Dis. 2019, 69, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tehrani, K.H.; Martin, N.I. β-lactam/β-lactamase inhibitor combinations: An update. Medchemcomm 2018, 9, 1439–1456. [Google Scholar] [CrossRef] [PubMed]
- Docquier, J.-D.; Lamotte-Brasseur, J.; Galleni, M.; Amicosante, G.; Frère, J.-M.; Rossolini, G.M. On functional and structural heterogeneity of VIM-type metallo-β-lactamases. J. Antimicrob. Chemother. 2003, 51, 257–266. [Google Scholar] [CrossRef]
- King, A.M.; Reid-Yu, S.A.; Wang, W.; King, D.T.; De Pascale, G.; Strynadka, N.C.; Walsh, T.R.; Coombes, B.K.; Wright, G.D. Aspergillomarasmine A overcomes metallo-β-lactamase antibiotic resistance. Nature 2014, 510, 503–506. [Google Scholar] [CrossRef] [Green Version]
- Sychantha, D.; Rotondo, C.M.; Tehrani, K.H.; Martin, N.I.; Wright, G.D. Aspergillomarasmine A inhibits metallo-β-lactamases by selectively sequestering Zn2+. J. Biol. Chem. 2021, 297, 100918. [Google Scholar] [CrossRef]
- Samuelsen, Ø.; Åstrand, O.A.H.; Fröhlich, C.; Heikal, A.; Skagseth, S.; Carlsen, T.J.O.; Leiros, H.-K.S.; Bayer, A.; Schnaars, C.; Kildahl-Andersen, G. ZN148 is a modular synthetic metallo-β-lactamase inhibitor that reverses carbapenem resistance in Gram-negative pathogens In Vivo. Antimicrob. Agents Chemother. 2020, 64, e02415-19. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Bai, M.; Liu, W.; Kong, H.; Zhang, T.; Yao, H.; Zhang, E.; Du, J.; Qin, S. H2dpa derivatives containing pentadentate ligands: An acyclic adjuvant potentiates meropenem activity in vitro and in vivo against metallo-β-lactamase-producing Enterobacterales. Eur. J. Med. Chem. 2021, 224, 113702. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.W.; Cho, E.J.; Bethel, C.R.; Smisek, T.; Ahn, Y.-C.; Schroeder, J.M.; Thomas, C.A.; Dalby, K.N.; Beckham, J.T.; Crowder, M.W. Discovery of an effective small-molecule allosteric inhibitor of New Delhi metallo-β-lactamase (NDM). Acs Infect. Dis. 2022, 8, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Zalacain, M.; Lozano, C.; Llanos, A.; Sprynski, N.; Valmont, T.; De Piano, C.; Davies, D.; Leiris, S.; Sable, C.; Ledoux, A. Novel specific metallo-β-lactamase inhibitor ANT2681 restores meropenem activity to clinically effective levels against NDM-positive Enterobacterales. Antimicrob. Agents Chemother. 2021, 65, e00203-21. [Google Scholar] [CrossRef] [PubMed]
- Farley, A.J.; Ermolovich, Y.; Calvopiña, K.; Rabe, P.; Panduwawala, T.; Brem, J.r.; Bjorkling, F.; Schofield, C.J. Structural Basis of Metallo-β-lactamase Inhibition by N-Sulfamoylpyrrole-2-carboxylates. Acs Infect. Dis. 2021, 7, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Legru, A.; Verdirosa, F.; Hernandez, J.-F.; Tassone, G.; Sannio, F.; Benvenuti, M.; Conde, P.-A.; Bossis, G.; Thomas, C.A.; Crowder, M.W. 1,2,4-Triazole-3-thione compounds with a 4-ethyl alkyl/aryl sulfide substituent are broad-spectrum metallo-β-lactamase inhibitors with re-sensitization activity. Eur. J. Med. Chem. 2021, 226, 113873. [Google Scholar] [CrossRef]
- Shi, C.; Chen, J.; Kang, X.; Shen, X.; Lao, X.; Zheng, H. Approaches for the discovery of metallo-β-lactamase inhibitors: A review. Chem. Biol. Drug Des. 2019, 94, 1427–1440. [Google Scholar] [CrossRef]
- Somboro, A.M.; Tiwari, D.; Bester, L.A.; Parboosing, R.; Chonco, L.; Kruger, H.G.; Arvidsson, P.I.; Govender, T.; Naicker, T.; Essack, S.Y. NOTA: A potent metallo-β-lactamase inhibitor. J. Antimicrob. Chemother. 2015, 70, 1594–1596. [Google Scholar] [CrossRef] [Green Version]
- Azumah, R.; Dutta, J.; Somboro, A.; Ramtahal, M.; Chonco, L.; Parboosing, R.; Bester, L.; Kruger, H.; Naicker, T.; Essack, S. In vitro evaluation of metal chelators as potential metallo-β-lactamase inhibitors. J. Appl. Microbiol. 2016, 120, 860–867. [Google Scholar] [CrossRef] [Green Version]
- Omolabi, K.F.; Reddy, N.; Mdanda, S.; Ntshangase, S.; Singh, S.D.; Kruger, H.G.; Naicker, T.; Govender, T.; Bajinath, S. The in vitro and in vivo potential of metal-chelating agents as metallo-beta-lactamase inhibitors against carbapenem-resistant Enterobacterales. Fems Microbiol. Lett. 2022, 370, fnac122. [Google Scholar] [CrossRef]
- Peters, B.K.; Reddy, N.; Shungube, M.; Girdhari, L.; Baijnath, S.; Mdanda, S.; Chetty, L.; Ntombela, T.; Arumugam, T.; Bester, L.A.; et al. The in vitro and in vivo development of a β-lactam-metallo-β-lactamase inhibitor: Targeting carbapenem-resistant Enterobacterales. Acs Infect. Dis. 2023, 9, 486–496. [Google Scholar] [CrossRef]
- Reller, L.B.; Weinstein, M.; Jorgensen, J.H.; Ferraro, M.J. Antimicrobial susceptibility testing: A review of general principles and contemporary practices. Clin. Infect. Dis. 2009, 49, 1749–1755. [Google Scholar] [CrossRef]
- Mouton, J.W.; Muller, A.E.; Canton, R.; Giske, C.G.; Kahlmeter, G.; Turnidge, J. MIC-based dose adjustment: Facts and fables. J. Antimicrob. Chemother. 2018, 73, 564–568. [Google Scholar] [CrossRef] [Green Version]
- Giraldo, J.; Serra, J.; Roche, D.; Rovira, X. Assessing receptor affinity for inverse agonists: Schild and Cheng-Prusoff methods revisited. Curr. Drug Targets 2007, 8, 197–202. [Google Scholar] [CrossRef]
- Wade, N.; Tehrani, K.H.; Brüchle, N.C.; van Haren, M.J.; Mashayekhi, V.; Martin, N.I. Mechanistic investigations of metallo-β-lactamase inhibitors: Strong zinc binding Is not required for potent enzyme inhibition. ChemMedChem 2021, 16, 1651–1659. [Google Scholar] [CrossRef]
- Daiyasu, H.; Osaka, K.; Ishino, Y.; Toh, H. Expansion of the zinc metallo-hydrolase family of the β-lactamase fold. Febs Lett. 2001, 503, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asempa, T.E.; Abdelraouf, K.; Nicolau, D.P. Activity of β-lactam antibiotics against metallo-β-lactamase-producing Enterobacterales in animal infection models: A current state of affairs. Antimicrob. Agents Chemother. 2021, 65, e02271-20. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Reuter, S.; Harris, S.R.; Glasner, C.; Feltwell, T.; Argimon, S.; Abudahab, K.; Goater, R.; Giani, T.; Errico, G. Epidemic of carbapenem-resistant Klebsiella pneumoniae in Europe is driven by nosocomial spread. Nat. Microbiol. 2019, 4, 1919–1929. [Google Scholar] [CrossRef]
- Everett, M.; Sprynski, N.; Coelho, A.; Castandet, J.; Bayet, M.; Bougnon, J.; Lozano, C.; Davies, D.T.; Leiris, S.; Zalacain, M. Discovery of a novel metallo-β-lactamase inhibitor that potentiates meropenem activity against carbapenem-resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62, e00074-18. [Google Scholar] [CrossRef] [Green Version]
- Schnaars, C.; Kildahl-Andersen, G.; Prandina, A.; Popal, R.; Radix, S.; Le Borgne, M.; Gjøen, T.; Andresen, A.M.S.; Heikal, A.; Økstad, O.A. Synthesis and preclinical evaluation of TPA-based zinc chelators as metallo-β-lactamase inhibitors. Acs Infect. Dis. 2018, 4, 1407–1422. [Google Scholar] [CrossRef]
- Kildahl-Andersen, G.; Schnaars, C.; Prandina, A.; Radix, S.; Le Borgne, M.; Jordheim, L.P.; Gjøen, T.; Andresen, A.M.S.; Lauksund, S.; Fröhlich, C. Synthesis and biological evaluation of zinc chelating compounds as metallo-β-lactamase inhibitors. Medchemcomm 2019, 10, 528–537. [Google Scholar] [CrossRef]
- Prandina, A.; Radix, S.; Le Borgne, M.; Jordheim, L.P.; Bousfiha, Z.; Fröhlich, C.; Leiros, H.-K.S.; Samuelsen, Ø.; Frøvold, E.; Rongved, P. Synthesis and biological evaluation of new dipicolylamine zinc chelators as metallo-β-lactamase inhibitors. Tetrahedron 2019, 75, 1525–1540. [Google Scholar] [CrossRef]
- Ishii, Y.; Eto, M.; Mano, Y.; Tateda, K.; Yamaguchi, K. In vitro potentiation of carbapenems with ME1071, a novel metallo-β-lactamase inhibitor, against metallo-β-lactamase-producing Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 2010, 54, 3625–3629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosibo, S.C.; Somboro, A.M.; Amoako, D.G.; Osei Sekyere, J.; Bester, L.A.; Ngila, J.C.; Sun, D.D.; Kumalo, H.M. Impact of Pyridyl Moieties on the Inhibitory Properties of Prominent Acyclic Metal Chelators against Metallo-β-Lactamase-Producing Enterobacteriaceae: Investigating the Molecular Basis of Acyclic Metal Chelators’ Activity. Microb. Drug Resist. 2019, 25, 439–449. [Google Scholar] [CrossRef]
- Harada, R.; Kitao, A. Parallel cascade selection molecular dynamics (PaCS-MD) to generate conformational transition pathway. J. Chem. Phys. 2013, 139, 035103. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Zou, Y.; Zhan, M.; Guo, Q.; Zhang, Y.; Zhang, Z.; Li, B.; Zhang, S.; Chu, H. Zinc Chelator N,N,N′,N′-Tetrakis (2-Pyridylmethyl) Ethylenediamine Reduces the Resistance of Mycobacterium abscessus to Imipenem. Infect. Drug Resist. 2020, 13, 2883–2890. [Google Scholar] [CrossRef]
- Nordmann, P.; Poirel, L.; Dortet, L. Rapid detection of carbapenemase-producing Enterobacteriaceae. Emerg. Infect. Dis. 2012, 18, 1503–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacVane, S.H.; Crandon, J.L.; Nichols, W.W.; Nicolau, D.P. In vivo efficacy of humanized exposures of ceftazidime-avibactam in comparison with ceftazidime against contemporary Enterobacteriaceae isolates. Antimicrob. Agents Chemother. 2014, 58, 6913–6919. [Google Scholar] [CrossRef] [Green Version]
- CALIS-C. Performance Standards for Antimicrobial Susceptibility Testing: Approved Twenty: Document M100-S28; CLSI: Wayne, PA, USA, 2018; Volume 2018. [Google Scholar]
- Hsieh, M.H.; Chen, M.Y.; Victor, L.Y.; Chow, J.W. Synergy assessed by checkerboard a critical analysis. Diagn. Microbiol. Infect. Dis. 1993, 16, 343–349. [Google Scholar] [CrossRef]
- Bardbari, A.M.; Arabestani, M.R.; Karami, M.; Keramat, F.; Aghazadeh, H.; Alikhani, M.Y.; Bagheri, K.P. Highly synergistic activity of melittin with imipenem and colistin in biofilm inhibition against multidrug-resistant strong biofilm producer strains of Acinetobacter baumannii. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 443–454. [Google Scholar] [CrossRef]
- Giacometti, A.; Cirioni, O.; Kamysz, W.; D’Amato, G.; Silvestri, C.; Del Prete, M.S.; Łukasiak, J.; Scalise, G. Comparative activities of cecropin A, melittin, and cecropin A–melittin peptide CA (1–7) M (2–9) NH2 against multidrug-resistant nosocomial isolates of Acinetobacter baumannii. Peptides 2003, 24, 1315–1318. [Google Scholar] [CrossRef]
- CaLSI-C. Methods for Determining Bacterial Activity of Antimicrobial Agents; Approved Guideline: M26A.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 1999. [Google Scholar]
- Michail, G.; Labrou, M.; Pitiriga, V.; Manousaka, S.; Sakellaridis, N.; Tsakris, A.; Pournaras, S. Activity of tigecycline in combination with colistin, meropenem, rifampin, or gentamicin against KPC-producing Enterobacteriaceae in a murine thigh infection model. Antimicrob. Agents Chemother. 2013, 57, 6028–6033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, B.K.; Kruger, H.G.; Arvidsson, P.I.; Naicker, T.; Govender, T. Metallo-Beta-Lactamase Inhibitors. International Patent PCT/IB2022/056748, 21 July 2023. [Google Scholar]
- Hu, D.X.; Grice, P.; Ley, S.V. Rotamers or Diastereomers? An Overlooked NMR Solution. J. Org. Chem. 2012, 77, 5198–5202. [Google Scholar] [CrossRef]
- Petzoldlab. Available online: http://petzoldlab.com (accessed on 1 April 2022).
- Akitt, J.W. N.M.R. and Chemistry; Chapman and Hall Ltd. CUP Archive: London, UK, 1973. [Google Scholar]
- Christopeit, T.; Yang, K.-W.; Yang, S.-K.; Leiros, H.-K.S. The structure of the metallo-β-lactamase VIM-2 in complex with a triazolylthioacetamide inhibitor. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 813–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Ding, J.; Zhu, D.; Liu, X.; Xu, X.; Zhang, Y.; Zang, S.; Wang, D.-C.; Liu, W. Structural and mechanistic insights into NDM-1 catalyzed hydrolysis of cephalosporins. J. Am. Chem. Soc. 2014, 136, 14694–14697. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Theor. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Theor. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins 2005, 61, 704–721. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D. OPLS4: Improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Release, S. 3: Desmond Molecular Dynamics System; DE Shaw Research: New York, NY, USA, 2017. [Google Scholar]
- Price, D.J.; Brooks III, C.L. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Reference | Bacterial Strain | MBL Produced | MIC (mg/L) | FICI | ||
|---|---|---|---|---|---|---|
| MEM Alone | BP2 Alone | BP2 + MEM | ||||
| ATCC 25922 | Escherichia coli | N/A | 0.06 | N/A | 0 + 0.06 | N/A |
| AUS 271 | Escherichia coli | NDM-1 | >128 | >256 | 16 + 0.25 | 0.13 |
| FEK | Escherichia coli | NDM-4 | >32 | >256 | 16 + 0.5 | 0.07 |
| JAP | Escherichia coli | IMP-1 | 8 | >256 | 16 + 0.03 | 0.04 |
| TWA | Escherichia coli | IMP-8 | 8 | >256 | 32 + 0.06 | 0.09 |
| BM 14 | Escherichia coli | VIM-1 | 32 | >256 | 8 + 0.25 | 0.04 |
| TC CARF | Escherichia coli | VIM-2 | 8 | >256 | 4 + 0.5 | 0.08 |
| DIH | Escherichia coli | VIM-19 | >32 | >256 | 16 + 0.125 | 0.07 |
| IR 386 | Enterobacter cloacae | NDM-1 | 32 | >256 | 16 + 0.125 | 0.07 |
| BM 5 | Enterobacter cloacae | IMP-1 | >32 | >256 | 32 + 0.5 | 0.14 |
| TWA | Enterobacter cloacae | IMP-8 | 0.125 | >256 | 16 + 0.06 | 0.5 |
| KAR | Enterobacter cloacae | VIM-1 | 4 | >256 | 16 + 0.25 | 0.13 |
| USA 449 | Klebsiella pneumoniae | NDM | >32 | >256 | 16 + 0.25 | 0.07 |
| 6852 | Klebsiella pneumoniae | IMP-1 | >32 | >256 | 64 + 1 | 0.3 |
| TWA | Klebsiella pneumoniae | IMP-8 | 8 | >256 | 16 + 0.25 | 0.09 |
| ENNES | Klebsiella pneumoniae | VIM-1 | >32 | >256 | 8 + 0.5 | 0.05 |
| DIH | Klebsiella pneumoniae | VIM-19 | 32 | >256 | 16 + 0.125 | 0.07 |
| Serratia marcescens | IMP-11 | >32 | >256 | 64 + 1 | 0.3 | |
| BM 20 | Serratia marcescens | VIM-2 | >32 | >256 | 8 + 0.5 | 0.05 |
| PSTU | Providencia stuartii | NDM-1 | 32 | >256 | 8 + 0.25 | 0.04 |
| IR38 | Providencia Rettgeri | NDM-1 | 32 | >256 | 32 + 0.5 | 0.14 |
| MBL | IC50 (µM) | Ki app (µM) |
|---|---|---|
| NDM-1 | 70.7 ± 4.5 | 35.3 |
| VIM-2 | 57.1 ± 2.9 | 30.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, N.; Girdhari, L.; Shungube, M.; Gouws, A.C.; Peters, B.K.; Rajbongshi, K.K.; Baijnath, S.; Mdanda, S.; Ntombela, T.; Arumugam, T.; et al. Neutralizing Carbapenem Resistance by Co-Administering Meropenem with Novel β-Lactam-Metallo-β-Lactamase Inhibitors. Antibiotics 2023, 12, 633. https://doi.org/10.3390/antibiotics12040633
Reddy N, Girdhari L, Shungube M, Gouws AC, Peters BK, Rajbongshi KK, Baijnath S, Mdanda S, Ntombela T, Arumugam T, et al. Neutralizing Carbapenem Resistance by Co-Administering Meropenem with Novel β-Lactam-Metallo-β-Lactamase Inhibitors. Antibiotics. 2023; 12(4):633. https://doi.org/10.3390/antibiotics12040633
Chicago/Turabian StyleReddy, Nakita, Letisha Girdhari, Mbongeni Shungube, Arnoldus C. Gouws, Byron K. Peters, Kamal K. Rajbongshi, Sooraj Baijnath, Sipho Mdanda, Thandokuhle Ntombela, Thilona Arumugam, and et al. 2023. "Neutralizing Carbapenem Resistance by Co-Administering Meropenem with Novel β-Lactam-Metallo-β-Lactamase Inhibitors" Antibiotics 12, no. 4: 633. https://doi.org/10.3390/antibiotics12040633