
Multiresidues Multiclass Analytical Methods for Determination of Antibiotics in Animal Origin Food: A Critical Analysis

Abstract
:1. Introduction
2. Legal Frame of Maximum Residue Limits (MRL) of Antibiotics in Foods of Animal Origin
3. Physico-Chemical Properties of Different Classes of Antibiotics
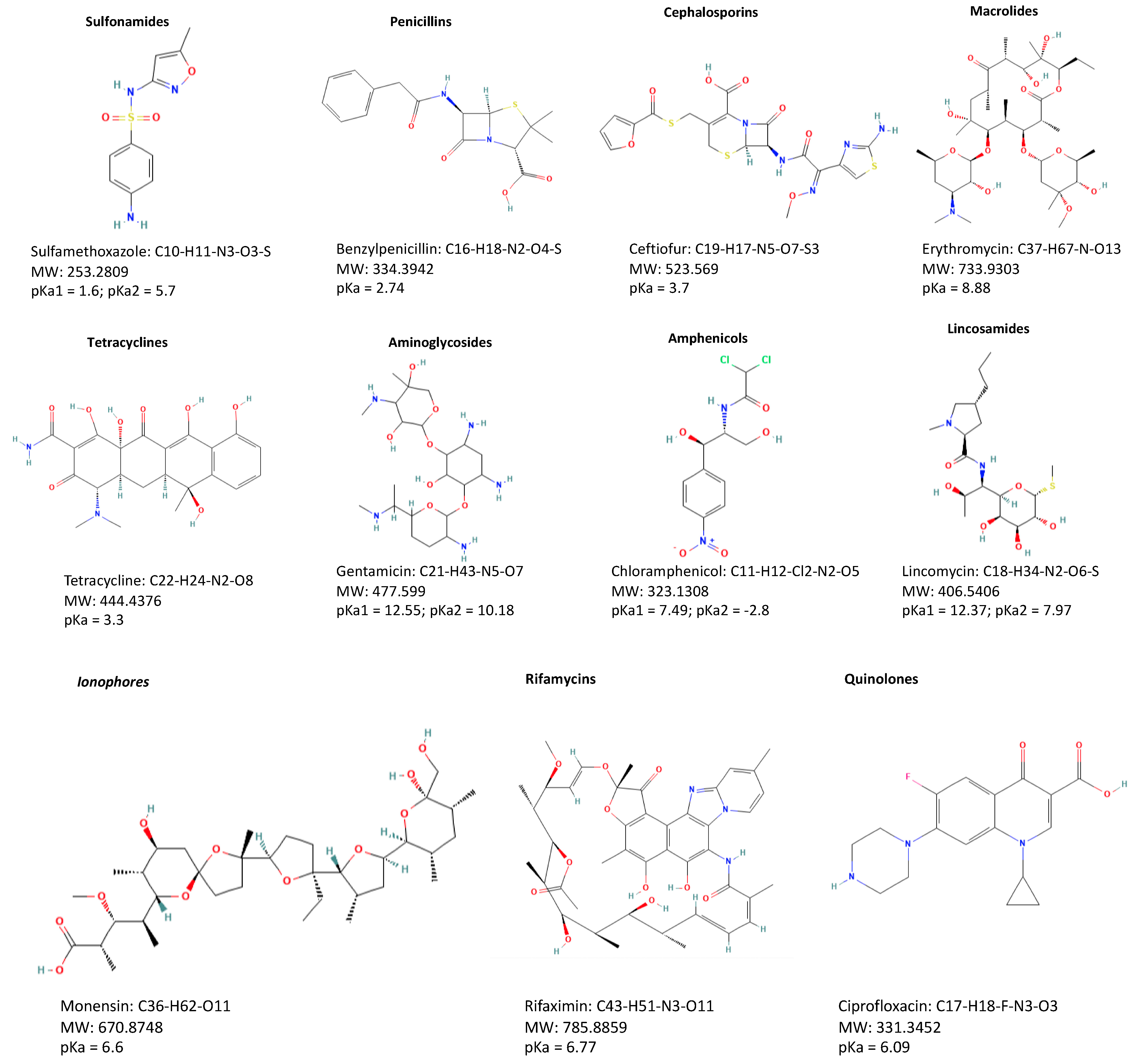
3.1. Sulfonamides
3.2. Tetracyclines
3.3. Penicillins
3.4. Cephalosporins
3.5. Macrolides
3.6. Quinolones, including Fluoroquinolones
3.7. Aminoglycosides
3.8. Phenicols
3.9. Lincosamides
3.10. Polymyxins
3.11. Bacitracins
3.12. Novobiocin
3.13. Tiamulin
3.14. Ionophores
3.15. Rifamycins
4. Extraction and Clean-Up Methods for the Determination of Antibiotic Residues
5. Multiclass Multiresidue Methods
5.1. Detection Methods for Multiresidues of Antibiotics
- Chromatographic methods: from HPLC to UHPLC
- Chromatographic methods: analytical columns
- Chromatographic methods: the selection of the mobile phase
- Chromatographic methods: from tandem mass spectrometry to high resolution mass spectrometry
5.2. Non-Chromatographic Methods
6. Effect of Food Processing on the Residues of Antibiotics Found in Animal Products
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| AG | Aminoglycosides |
| AM | Amphenicols |
| AMX | Amoxicillin |
| AR | Antibiotics residues |
| AT | Anthelmintics |
| AV | Avermectins |
| BA | β-Agonists |
| BZM | Benzimidazole |
| CAP | Chloramphenicol |
| CC alfa | Decision limit |
| CC beta | Detection capability |
| CCD | Coccidiostats |
| CDC | Glucocorticoids |
| CL | Chemiluminescence |
| CP | cephalosporines |
| CTC | Chlortetracycline |
| Cval | Validation levels |
| DC | Doxycycline |
| dDMIP | Dummy molecularly imprinted polymer |
| DP | Diaminopyrimidines |
| DPX | weak cation exchange cartridges |
| dSPE | Solid-phase dispersion |
| d-SPME | Dispersive solid-phase microextraction |
| DYE | Dyes |
| EAEU | Eurasian Economic Union |
| EEA | European Economic Area |
| EMR-L | Enhanced Matrix Removal for Lipid |
| EFSA | European Food Safety Authority |
| ELISA | Enzyme-Linked Immune Sorbent Assay |
| EDTA | Ethylenediaminetetraacetic acid |
| EU | European Union |
| FA | Formic Acid |
| FDA | Food and Drug Administration |
| FF | Florfenicol |
| FVO | Food and Veterinary Office |
| GCC | Gulf Cooperation Council |
| HCL | Hydrochloric acid |
| HFBA | Heptafluorobutyric acid |
| HILIC–MS/MS | Hydrophilic Interaction Liquid Chromatography tandem Mass Spectrometry |
| HLB | Hydrophilic-lipophilic balance |
| HLB PRiME | Hydrophilic-lipophilic balance PRiME (process, robustness, improvements, matrix effects, ease of use) |
| HOM | Hormones |
| HPLC | High Performance Liquid Chromatography |
| HRMS | High-Resolution MS detectors |
| IO | Ionophores |
| IP | Ion Pairing |
| IZ | Imidazoles |
| LC | Liquid Chromatography |
| LC-MS/MS | Liquid Chromatography tandem Mass Spectrometry |
| LLE | Liquid-liquid extraction |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| LS | Lincosamides |
| MA | Macrolides |
| MAE | Microwave-assisted extraction |
| MEOH | Methanol |
| MEPS | Microextraction by packed sorbent |
| Micro-SPE MSPD | Micro-solid-phase extraction Matrix solid-phase dispersion |
| MIP | Molecularly imprinted polymer |
| MIP-SPE | Molecularly imprinted polymer solid-phase extraction |
| MISPE | Molecularly imprinted solid-phase extraction |
| MNPs | magnetic nanoparticles |
| MRL | Maximum residue limits |
| MS/MS | Tandem Mass Spectrometry |
| MSPE | Magnetic solid phase extraction |
| NMZ | Nitroimidazoles |
| OTC | Oxytetracycline |
| PDMS | Polydimethylsiloxane; |
| PEG | Poly ethylene glycol |
| PFPA | Pentafluoropropionic acid |
| PHWE | Pressurized hot water extraction |
| PLE | Pressurized liquid extraction |
| PN | Penicillins |
| PP | Polypeptides (Polymyxins) |
| PSA | Primary secondary amine |
| PT | Pleuromutilines |
| QDs | Quantum Dots |
| QN | Quinolones |
| QNX | Quinoxaline |
| QuEChERS | Quick, easy, cheap, effective, rugged, and safe |
| Q-Orbitrap | Quadrupole-Orbitrap mass spectrometer |
| QqQ | Triple quadrupole mass detector |
| QToF | Quadrupole Time-of-Flight mass spectrometer |
| RASFF | Rapid Alert System for Food and Feed |
| AS | Sulfonamides (including Dapsone) |
| SBSE | Stir-bar sorptive extraction |
| SDZ | Sulfadiazine |
| SL | Screening limit |
| SLE | Solid–Liquid Extraction |
| SMX | Sulfamethoxazole |
| SMZ | Sulfamethazine |
| SOSLE | Salting-out supported liquid extraction |
| SPE | Solid phase extraction |
| SPME | Solid-phase microextraction |
| SPR | Surface Plasmon Resonance |
| SQX | Sulfaquinoxaline |
| STC | Screening Target Concentration |
| TAP | Thiamphenicol |
| TC | Tetracyclines |
| TCA | Trichloroacetic acid |
| TFA | Trifluoroacetic acid |
| TQ | Tranquilizers |
| TT | Anti-thyroids |
| ToF | Time of Flight |
| UHPLC-QqQ | UHPLC coupled to triple quadrupole mass spectrometry |
| UHPLC | Ultra-High-Performance Liquid Chromatography |
| UAE | Ultrasonic-assisted extraction |
| UCNPs | Upward Converting Nanoparticles |
| UHPLC–MS/MS | Ultra-High-Performance Liquid Chromatography tandem Mass Spectrometry |
| UHPLC-Q/ToF | UHPLC coupled to Time of Flight mass spectrometry |
| VMPs | Veterinary Medicinal Products |
References
- Commission, E. Veterinary Medicines and Medicated Feed. Available online: https://food.ec.europa.eu/animals/animal-health/vet-meds-med-feed_en (accessed on 20 November 2022).
- Patel, S.J.; Wellington, M.; Shah, R.M.; Ferreira, M.J. Antibiotic Stewardship in Food-Producing Animals: Challenges, Progress, and Opportunities. Clin. Ther. 2020, 42, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- The European Parliament and the Council of the European. Union Regulation (EC) No 1831/2003 of the European Parliament and of the Council of 22 September 2003 on additives for use in animal nutrition. Off. J. Eur. Union 2003, 268, 29–43. [Google Scholar]
- Marazuela, M.D.; Bogialli, S. A review of novel strategies of sample preparation for the determination of antibacterial residues in foodstuffs using liquid chromatography-based analytical methods. Anal. Chim. Acta 2009, 645, 5–17. [Google Scholar] [CrossRef]
- Lechner, I.; Freivogel, C.; Stärk, K.D.C.; Visschers, V.H.M. Exposure Pathways to Antimicrobial Resistance at the Human-Animal Interface—A Qualitative Comparison of Swiss Expert and Consumer Opinions. Front. Public Health 2020, 8. [Google Scholar] [CrossRef]
- Kuppusamy, S.; Kakarla, D.; Venkateswarlu, K.; Megharaj, M.; Yoon, Y.-E.; Lee, Y.B. Veterinary antibiotics (VAs) contamination as a global agro-ecological issue: A critical view. Agric. Ecosyst. Environ. 2018, 257, 47–59. [Google Scholar] [CrossRef]
- Tirado, M.C.; Clarke, R.; Jaykus, L.A.; McQuatters-Gollop, A.; Frank, J.M. Climate change and food safety: A review. Food Res. Int. 2010, 43, 1745–1765. [Google Scholar] [CrossRef]
- Stavroulaki, A.; Tzatzarakis, M.N.; Karzi, V.; Katsikantami, I.; Renieri, E.; Vakonaki, E.; Avgenaki, M.; Alegakis, A.; Stan, M.; Kavvalakis, M.; et al. Antibiotics in Raw Meat Samples: Estimation of Dietary Exposure and Risk Assessment. Toxics 2022, 10, 456. [Google Scholar] [CrossRef]
- Mainero Rocca, L.; Gentili, A.; Pérez-Fernández, V.; Tomai, P. Veterinary drugs residues: A review of the latest analytical research on sample preparation and LC-MS based methods. Food Addit. Contam. Part A 2017, 34, 766–784. [Google Scholar] [CrossRef]
- The European Commission. Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. Off. J. Eur. Union. 2009, 15, 1–72. [Google Scholar]
- Cheng, C. Codex alimentarius. Encycl. Food Secur. Sustain. 1993, 31, 50–55. [Google Scholar]
- FDA. The Code of Federal Regulations, U.S.F.& D.A. TITLE 21--FOOD AND DRUGS. Available online: https://www.accessdata.fda.gov/ (accessed on 20 November 2022).
- World Health Organization. WHO Guidelines on Use of Medically Important Antimicrobials in Food-Producing Animals: Web Annex A: Evidence Base; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- European Commission (EC). Commission Decision 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Communities 2002, 221, 8–36. [Google Scholar]
- Rapid Alert System in Food and Feed [RASFF], 2019. Brussels European Commission. Available online: https://ec.europa.eu/food/safety/rasff_en (accessed on 20 November 2022).
- European Food Safety Authority. Report for 2019 on the results from the monitoring of veterinary medicinal product residues and other substances in live animals and animal products. EFSA Support. Publ. 2021, 18, 1–82. [Google Scholar]
- Veterinary Manual. Available online: https://www.merckvetmanual.com/pharmacology/antibacterial-agents (accessed on 20 November 2022).
- Remko, M.; von der Lieth, C.-W. Theoretical study of gas-phase acidity, pKa, lipophilicity, and solubility of some biologically active sulfonamides. Bioorg. Med. Chem. 2004, 12, 5395–5403. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, M.; Pellerano, R.G.; Pezza, L.; Pezza, H.R. An overview of the main foodstuff sample preparation technologies for tetracycline residue determination. Talanta 2018, 182, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Manimekalai, M.; Rawson, A.; Sengar, A.S.; Kumar, K.S. Development, Optimization, and Validation of Methods for Quantification of Veterinary Drug Residues in Complex Food Matrices Using Liquid-Chromatography—A Review. Food Anal. Methods 2019, 12, 1823–1837. [Google Scholar] [CrossRef]
- Lopes, R.P.; Reyes, R.C.; Romero-González, R.; Frenich, A.G.; Vidal, J.L.M. Development and validation of a multiclass method for the determination of veterinary drug residues in chicken by ultra high performance liquid chromatography–tandem mass spectrometry. Talanta 2012, 89, 201–208. [Google Scholar] [CrossRef]
- Rossi, R.; Saluti, G.; Moretti, S.; Diamanti, I.; Giusepponi, D.; Galarini, R. Multiclass methods for the analysis of antibiotic residues in milk by liquid chromatography coupled to mass spectrometry: A review. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2018, 35, 241–257. [Google Scholar] [CrossRef]
- Bessaire, T.; Mujahid, C.; Beck, A.; Tarres, A.; Savoy, M.C.; Woo, P.M.; Mottier, P.; Desmarchelier, A. Screening of 23 β-lactams in foodstuffs by LC–MS/MS using an alkaline QuEChERS-like extraction. Food Addit. Contam.-Part A Chem. Anal. Control Expo. Risk Assess. 2018, 35, 661–673. [Google Scholar] [CrossRef]
- Mazzei, T.; Mini, E.; Novelli, A.; Periti, P. Chemistry and mode of action of macrolides. J. Antimicrob. Chemother. 1993, 31, 1–9. [Google Scholar] [CrossRef]
- Stolker, A.A.M.; Brinkman, U.A.T. Analytical strategies for residue analysis of veterinary drugs and growth-promoting agents in food-producing animals–A review. J. Chromatogr. A 2005, 1067, 15–53. [Google Scholar] [CrossRef]
- Parshikov, I.A.; Sutherland, J.B. Microbial transformations of antimicrobial quinolones and related drugs. J. Ind. Microbiol. Biotechnol. 2012, 39, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Razzagh, M.; Reza, N. Determination of enrofloxacin residue in chicken eggs using FPT and ELISA methods. J. Res. Health 2015, 5, 159–164. [Google Scholar]
- Moghadam, N.R.; Arefhosseini, S.R.; Javadi, A.; Lotfipur, F.; Ansarin, M.; Tamizi, E.; Nemati, M. Determination of Enrofloxacin and Ciprofloxacin Residues in Five Different Kinds of Chicken Tissues by Dispersive Liquid-Liquid Microextraction Coupled with HPLC. Iran. J. Pharm. Res. 2018, 17, 1182–1190. [Google Scholar]
- Petrović, J.; Baltic, M.; Cupic, V.; Stefanović, S.; Stojanovic, D. Residues of enrofloxacin and its main metabolite ciprofloxacin in broiler chickens. Acta Vet. Brno. 2006, 56, 497–506. [Google Scholar]
- Pagkalis, S.; Mantadakis, E.; Mavros, M.N.; Ammari, C.; Falagas, M.E. Pharmacological Considerations for the Proper Clinical Use of Aminoglycosides. Drugs 2011, 71, 2277–2294. [Google Scholar] [CrossRef] [PubMed]
- Jank, L.; Martins, M.T.; Arsand, J.B.; Hoff, R.B.; Barreto, F.; Pizzolato, T.M.; Campos Motta, T.M. High-throughput method for macrolides and lincosamides antibiotics residues analysis in milk and muscle using a simple liquid-liquid extraction technique and liquid chromatography-electrospray-tandem mass spectrometry analysis (LC-MS/MS). Talanta 2015, 144, 686–695. [Google Scholar] [CrossRef]
- Tsuji, K.; Robertson, J.H. Improved high-performance liquid chromatographic method for polypeptide antibiotics and its application to study the effects of treatments to reduce microbial levels in bacitracin powder. J. Chromatogr. A 1975, 112, 663–672. [Google Scholar] [CrossRef]
- Brown, P.; Dawson, M.J. A Perspective on the Next Generation of Antibacterial Agents Derived by Manipulation of Natural Products. In Progress in Medicinal Chemistry; Lawton, G., Witty, D.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 54, pp. 135–184. ISBN 0079-6468. [Google Scholar]
- Maslow, M.J.; Portal-Celhay, C. Rifamycins. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier Saunders: Philadelphia, PA, USA, 2015; pp. 339–349.e3. ISBN 978-1-4557-4801-3. [Google Scholar]
- Anumol, T.; Lehotay, S.J.; Stevens, J.; Zweigenbaum, J. Comparison of veterinary drug residue results in animal tissues by ultrahigh-performance liquid chromatography coupled to triple quadrupole or quadrupole–time-of-flight tandem mass spectrometry after different sample preparation methods, including use of a commercial lipid removal product. Anal. Bioanal. Chem. 2017, 409, 2639–2653. [Google Scholar] [CrossRef]
- Jie, W.; Qiuhui, H.; Peng, L.; Yong, F.; Wenjian, Y.; Ning, M.; Fei, P. Comparison of three different lipid removal cleanup techniques prior to the analysis of sulfonamide drug residues in porcine tissues. Food Sci. Nutr. 2019, 7, 3006–3016. [Google Scholar]
- Bortolotte, A.R.; Daniel, D.; de Campos Braga, P.A.; Reyes, F.G.R. A simple and high-throughput method for multiresidue and multiclass quantitation of antimicrobials in pangasius (Pangasionodon hypophthalmus) fillet by liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1124, 17–25. [Google Scholar] [CrossRef]
- Magalhães, D.; Freitas, A.; Sofia Vila Pouca, A.; Barbosa, J.; Ramos, F. The use of ultra-high-pressure-liquid-chromatography tandem time-of-flight mass spectrometry as a confirmatory method in drug residue analysis: Application to the determination of antibiotics in piglet liver. J. Chromatogr. B 2020, 1153, 122264. [Google Scholar] [CrossRef] [PubMed]
- Kivrak, I.; Kivrak, S.; Harmandar, M. Development of a rapid method for the determination of antibiotic residues in honey using UPLC-ESI-MS/MS. FOOD Sci. Technol. 2016, 36, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, R.; Policastro, B.; Thomas, S.; Rice, D. Analysis and occurrence of 14 sulfonamide antibacterials and chloramphenicol in honey by solid-phase extraction followed by LC/MS/MS analysis. J. Agric. Food Chem. 2008, 56, 3509–3516. [Google Scholar] [CrossRef] [PubMed]
- Verzegnassi, L.; Savoy-Perroud, M.C.; Stadler, R.H. Application of liquid chromatography-electrospray ionization tandem mass spectrometry to the detection of 10 sulfonamides in honey. J. Chromatogr. A 2002, 977, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Grande-Martínez, Á.; Moreno-González, D.; Arrebola-Liébanas, F.J.; Garrido-Frenich, A.; García-Campaña, A.M. Optimization of a modified QuEChERS method for the determination of tetracyclines in fish muscle by UHPLC–MS/MS. J. Pharm. Biomed. Anal. 2018, 155, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Jansen, L.J.M.; Bolck, Y.J.C.; Rademaker, J.; Zuidema, T.; Berendsen, B.J.A. The analysis of tetracyclines, quinolones, macrolides, lincosamides, pleuromutilins, and sulfonamides in chicken feathers using UHPLC-MS/MS in order to monitor antibiotic use in the poultry sector. Anal. Bioanal. Chem. 2017, 409, 4927–4941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiesa, L.M.; Nobile, M.; Panseri, S.; Arioli, F. Antibiotic use in heavy pigs: Comparison between urine and muscle samples from food chain animals analysed by HPLC-MS/MS. Food Chem. 2017, 235, 111–118. [Google Scholar] [CrossRef]
- Reddy, B.; Sudhakar, Y.; Rao, Y.; Reddyprasad, P.; Sreedhar, N. LC-MS/MS Method, Development and Validation to Determine Three Tetracyclines and Their Epimers in Shrimp Samples. Orient. J. Chem. 2017, 33, 2459–2469. [Google Scholar] [CrossRef]
- Desmarchelier, A.; Anizan, S.; Minh Tien, M.; Savoy, M.-C.; Bion, C. Determination of five tetracyclines and their epimers by LC-MS/MS based on a liquid-liquid extraction with low temperature partitioning. Food Addit. Contam. Part A 2018, 35, 687–695. [Google Scholar] [CrossRef]
- Dasenaki, M.E.; Thomaidis, N.S. Multi-residue determination of 115 veterinary drugs and pharmaceutical residues in milk powder, butter, fish tissue and eggs using liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 2015, 880, 103–121. [Google Scholar] [CrossRef]
- Delatour, T.; Racault, L.; Bessaire, T.; Desmarchelier, A. Screening of veterinary drug residues in food by LC-MS/MS. Background and challenges. Food Addit. Contam.-Part A Chem. Anal. Control Expo. Risk Assess. 2018, 35, 632–645. [Google Scholar] [CrossRef] [PubMed]
- Yaqian, Z.; Xiang, L.; Xiaomao, L.; Jinjie, Z.; Yanzhong, C.; Zhihong, S.; Hanwen, S. Multi-class, multi-residue analysis of trace veterinary drugs in milk by rapid screening and quantification using ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry. J. Dairy Sci. 2015, 98, 8433–8444. [Google Scholar]
- Zheng, W.; Abd El-Aty, A.M.; Kim, S.K.; Choi, J.M.; Park, D.H.; Yoo, K.H.; Kang, Y.S.; Jeon, J.S.; Hacımüftüoğlu, A.; Shim, J.H.; et al. Development and validation of a solid-phase extraction method coupled with LC–MS/MS for the simultaneous determination of 16 antibiotic residues in duck meat. Biomed. Chromatogr. 2019, 33, e4501. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lucas, D.; Long, D.; Richter, B.; Stevens, J. Multi-class multi-residue analysis of veterinary drugs in meat using enhanced matrix removal lipid cleanup and liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2018, 1549, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Dubreil, E.; Gautier, S.; Fourmond, M.-P.; Bessiral, M.; Gaugain, M.; Verdon, E.; Pessel, D. Validation approach for a fast and simple targeted screening method for 75 antibiotics in meat and aquaculture products using LC-MS/MS. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2017, 34, 453–468. [Google Scholar] [CrossRef]
- Moats, W.A. Liquid Chromatographic Approaches to Determination of B-Lactam Antibiotic Residues in Milk and Tissues. In Analysis of Antibiotic/Drug Residues in Food Products of Animal Origin; Agarwal, V.K., Ed.; Springer US: Boston, MA, USA, 1992; pp. 133–145. ISBN 978-1-4615-3356-6. [Google Scholar]
- Van Holthoon, F.; Mulder, P.; Bennekom, E.; Heskamp, H.; Zuidema, T.; Rhijn, H. Quantitative analysis of penicillins in porcine tissues, milk and animal feed using derivatisation with piperidine and stable isotope dilution liquid chromatography tandem mass spectrometry. Anal. Bioanal. Chem. 2010, 396, 3027–3040. [Google Scholar] [CrossRef] [Green Version]
- Jank, L.; Hoff, R.B.; Tarouco, P.C.; Barreto, F.; Pizzolato, T.M. β-lactam antibiotics residues analysis in bovine milk by LC-ESI-MS/MS: A simple and fast liquid-liquid extraction method. Food Addit. Contam.-Part A Chem. Anal. Control Expo. Risk Assess. 2012, 29, 497–507. [Google Scholar] [CrossRef]
- Lara, F.J.; del Olmo-Iruela, M.; Cruces-Blanco, C.; Quesada-Molina, C.; Garcia-Campana, A.M. Advances in the determination of beta-lactam antibiotics by liquid chromatography. TRAC-TRENDS Anal. Chem. 2012, 38, 52–66. [Google Scholar] [CrossRef]
- Jank, L.; Martins, M.T.; Arsand, J.B.; Hoff, R.B.; Barreto, F.; Pizzolato, T.M. High-throughput method for the determination of residues of β-lactam antibiotics in bovine milk by LC-MS/MS. Food Addit. Contam.-Part A Chem. Anal. Control Expo. Risk Assess 2015, 32, 1992–2001. [Google Scholar] [CrossRef]
- Sun, L.; Jia, L.; Xie, X.; Xie, K.; Wang, J.; Liu, J.; Cui, L.; Zhang, G.; Dai, G.; Wang, J. Quantitative analysis of amoxicillin, its major metabolites and ampicillin in eggs by liquid chromatography combined with electrospray ionization tandem mass spectrometry. Food Chem. 2016, 192, 313–318. [Google Scholar] [CrossRef]
- Thompson, T.S.; van den Heever, J.P. Degradation of erythromycin in honey and selection of suitable marker residues for food safety analysis. Food Chem. 2012, 133, 1510–1520. [Google Scholar] [CrossRef]
- Salikin, J.; Abdullah, A. Determination of macrolide antibiotics in chicken tissues by liquid chromatography-electrospray mass spectrometry. AIP Conf. Proc. 2013, 1571, 702–709. [Google Scholar]
- Lan, C.; Yin, D.; Yang, Z.; Zhao, W.; Chen, Y.; Zhang, W.; Zhang, S. Determination of Six Macrolide Antibiotics in Chicken Sample by Liquid Chromatography-Tandem Mass Spectrometry Based on Solid Phase Extraction. J. Anal. Methods Chem. 2019, 2019, 6849457. [Google Scholar] [CrossRef] [Green Version]
- Benetti, C.; Piro, R.; Binato, G.; Angeletti, R.; Biancotto, G. Simultaneous determination of lincomycin and five macrolide antibiotic residues in honey by liquid chromatography coupled to electrospray ionization mass spectrometry (LC-MS/MS). Food Addit. Contam. 2006, 23, 1099–1108. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, Y.; Zhou, Y.; Liu, Z.; Meng, Q.; Feng, X. A review of pretreatment and analysis of macrolides in food (Update Since 2010). J. Chromatogr. A 2020, 1634, 461662. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Deng, F.; He, R.; Tan, L.; Luo, X.; Pan, X.; Yang, Z. A pass-through solid-phase extraction clean-up method for the determination of 11 quinolone antibiotics in chicken meat and egg samples using ultra-performance liquid chromatography tandem mass spectrometry. Microchem. J. 2019, 151, 104213. [Google Scholar] [CrossRef]
- Hassouan, M.; Ballesteros, O.; Taoufiki, J.; Vílchez, J.; Cabrera-Aguilera, M.; Navalón, A. Multiresidue determination of quinolone antibacterials in eggs of laying hens by liquid chromatography with fluorescence detection. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2007, 852, 625–630. [Google Scholar] [CrossRef]
- Bailac, S.; Barron, D.; Barbosa, J. New extraction procedure to improve the determination of quinolones in poultry muscle by liquid chromatography with ultraviolet and mass spectrometric detection. Anal. Chim. Acta 2006, 580, 163–169. [Google Scholar] [CrossRef]
- Yorke, J.C.; Froc, P. Quantitation of nine quinolones in chicken tissues by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. A 2000, 882, 63–77. [Google Scholar] [CrossRef]
- He, J.; Song, L.; Zhou, G.; Zhao, L. The Rapid Analysis of Antibiotics in Animal Meat and Egg Using a Novel SEP Method and UPLC–MS/MS. Chromatographia 2017, 80, 1329–1342. [Google Scholar] [CrossRef]
- Annunziata, L.; Visciano, P.; Stramenga, A.; Colagrande, M.; Campana, G.; Scortichini, G.; Migliorati, G.; Compagnone, D. Development and Validation of a Method for the Determination of Quinolones in Muscle and Eggs by Liquid Chromatography-Tandem Mass Spectrometry. Food Anal. Methods 2016, 9, 2308–2320. [Google Scholar] [CrossRef]
- Martínez-Villalba, A.; Moyano, E.; Galceran, M.T. Ultra-high performance liquid chromatography–atmospheric pressure chemical ionization–tandem mass spectrometry for the analysis of benzimidazole compounds in milk samples. J. Chromatogr. A 2013, 1313, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Liu, G.; Wang, F.; Sasanya, J.J.; Cannavan, A. Development of a Liquid Chromatography Tandem Mass Spectrometric Method for Simultaneous Determination of 15 Aminoglycoside Residues in Porcine Tissues. Food Anal. Methods 2016, 9, 2587–2599. [Google Scholar] [CrossRef]
- Dasenaki, M.E.; Michali, C.S.; Thomaidis, N.S. Analysis of 76 veterinary pharmaceuticals from 13 classes including aminoglycosides in bovine muscle by hydrophilic interaction liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2016, 1452, 67–80. [Google Scholar] [CrossRef]
- Bixia, Y.; Lian, W.; Chunying, L.U.O.; Xixi, W.; Chengjun, S.U.N. Simultaneous Determination of 11 Aminoglycoside Residues in Honey, Milk, and Pork by Liquid Chromatography with Tandem Mass Spectrometry and Molecularly Imprinted Polymer Solid Phase Extraction. J. AOAC Int. 2017, 100, 1869–1878. [Google Scholar]
- Kang, J.W.; Park, S.J.; Park, H.C.; Hossain, M.A.; Kim, M.A.; Son, S.W.; Lim, C.M.; Kim, T.W.; Cho, B.H. Multiresidue Screening of Veterinary Drugs in Meat, Milk, Egg, and Fish Using Liquid Chromatography Coupled with Ion Trap Time-of-Flight Mass Spectrometry. Appl. Biochem. Biotechnol. 2017, 182, 635–652. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Zhan, J.; Shi, X.; Deng, X.; Zhu, J.; Wu, W.; Chen, X. Analysis of 140 Veterinary Drugs and Other Contaminants in Poultry Muscle by Ultrahigh-Performance Liquid Chromatography–Tandem Mass Spectrometry. Chromatographia 2018, 81, 707–718. [Google Scholar] [CrossRef]
- Guidi, L.R.; Tette, P.A.S.; Gloria, M.B.A.; Fernandes, C. A simple and rapid LC–MS/MS method for the determination of amphenicols in Nile tilapia. Food Chem. 2018, 262, 235–241. [Google Scholar] [CrossRef]
- Lehotay, S.J.; Lightfield, A.R. Simultaneous analysis of aminoglycosides with many other classes of drug residues in bovine tissues by ultrahigh-performance liquid chromatography-tandem mass spectrometry using an ion-pairing reagent added to final extracts. Anal. Bioanal. Chem. 2018, 410, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Contin, M.D.; Quinsaat, J.E.; Negri, R.M.; Tripodi, V.P.; Opris, D.; D’Accorso, N.B. Development of carbohydrate functionalized magnetic nanoparticles for aminoglycosides magnetic solid phase extraction. Anal. Chim. Acta 2019, 1082, 37–48. [Google Scholar] [CrossRef]
- Castilla-Fernández, D.; Moreno-González, D.; Beneito-Cambra, M.; Molina-Díaz, A. Critical assessment of two sample treatment methods for multiresidue determination of veterinary drugs in milk by UHPLC-MS/MS. Anal. Bioanal. Chem. 2019, 411, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chi, Q.; Xia, S.; Pan, Y.; Chen, Y.; Wang, K. Untargeted multi-residue method for the simultaneous determination of 141 veterinary drugs and their metabolites in pork by high-performance liquid chromatography time-of-flight mass spectrometry. J. Chromatogr. A 2020, 1634, 461671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zeng, J.; Xiong, W.; Zeng, Z. Rapid determination of amoxicillin in porcine tissues by UPLC-MS/MS with internal standard. J. Food Compos. Anal. 2020, 92, 103578. [Google Scholar] [CrossRef]
- Glinka, M.; Wojnowski, W.; Wasik, A. Determination of aminoglycoside antibiotics: Current status and future trends. TrAC Trends Anal. Chem. 2020, 131, 116034. [Google Scholar] [CrossRef]
- Liu, Q.; Li, J.; Song, X.; Zhang, M.; Li, E.; Gao, F.; He, L. Simultaneous determination of aminoglycoside antibiotics in feeds using high performance liquid chromatography with evaporative light scattering detection. RSC Adv. 2017, 7, 1251–1259. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Leung, D. The challenges of developing a generic extraction procedure to analyze multi-class veterinary drug residues in milk and honey using ultra-high pressure liquid chromatography quadrupole time-of-flight mass spectrometry. Drug Test. Anal. 2012, 4, 103–111. [Google Scholar] [CrossRef]
- Guidi, L.R.; Tette, P.A.S.; Fernandes, C.; Silva, L.H.M.; Gloria, M.B.A. Advances on the chromatographic determination of amphenicols in food. Talanta 2017, 162, 324–338. [Google Scholar] [CrossRef]
- Giusepponi, D.; Paoletti, F.; Barola, C.; Moretti, S.; Saluti, G.; Galarini, R.; Ianni, F.; Sardella, R. Transfer of a multiclass method for over 60 antibiotics in food from high resolution to low resolution mass spectrometry. Molecules 2019, 24, 2935. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, A.; Widmer, M. Quantitative analysis of polypeptide antibiotic residues in a variety of food matrices by liquid chromatography coupled to tandem mass spectrometry. Anal. Chim. Acta 2013, 797, 81–88. [Google Scholar] [CrossRef]
- Saluti, G.; Diamanti, I.; Giusepponi, D.; Pucciarini, L.; Rossi, R.; Moretti, S.; Sardella, R.; Galarini, R. Simultaneous determination of aminoglycosides and colistins in food. Food Chem. 2018, 266, 9–16. [Google Scholar] [CrossRef]
- Schlüsener, M.P.; Bester, K.; Spiteller, M. Determination of antibiotics such as macrolides, ionophores and tiamulin in liquid manure by HPLC–MS/MS. Anal. Bioanal. Chem. 2003, 375, 942–947. [Google Scholar] [CrossRef] [PubMed]
- De Baere, S.; Devreese, M.; Maes, A.; De Backer, P.; Croubels, S. Quantification of 8-α-hydroxy-mutilin as marker residue for tiamulin in rabbit tissues by high-performance liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 4437–4445. [Google Scholar] [CrossRef]
- Tarannum, N.; Khatoon, S.; Dzantiev, B.B. Perspective and application of molecular imprinting approach for antibiotic detection in food and environmental samples: A critical review. Food Control 2020, 118, 107381. [Google Scholar] [CrossRef]
- Bitas, D.; Kabir, A.; Locatelli, M.; Samanidou, V. Food sample preparation for the determination of sulfonamides by high-performance liquid chromatography: State-of-the-Art. Separations 2018, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Khatibi, S.A.; Hamidi, S.; Siahi-Shadbad, M.R. Current trends in sample preparation by solid-phase extraction techniques for the determination of antibiotic residues in foodstuffs: A review. Crit. Rev. Food Sci. Nutr. 2020, 61, 3361–3382. [Google Scholar] [CrossRef]
- Dmitrienko, S.G.; Kochuk, E.V.; Apyari, V.V.; Tolmacheva, V.V.; Zolotov, Y.A. Recent advances in sample preparation techniques and methods of sulfonamides detection–A review. Anal. Chim. Acta 2014, 850, 6–25. [Google Scholar] [CrossRef] [PubMed]
- Moga, A.; Vergara-Barberan, M.; Lerma-Garcia, M.J.; Carrasco-Correa, E.J.; Herrero-Martinez, J.M.; Simo-Alfonso, E.F. Determination of antibiotics in meat samples using analytical methodologies: A review. Compr. Rev. Food Sci. Food Saf. 2021, 20, 1681–1716. [Google Scholar] [CrossRef]
- Chen, D.; Yu, J.; Tao, Y.; Pan, Y.; Xie, S.; Huang, L.; Peng, D.; Wang, X.; Wang, Y.; Liu, Z.; et al. Qualitative screening of veterinary anti-microbial agents in tissues, milk, and eggs of food-producing animals using liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. B 2016, 1017–1018, 82–88. [Google Scholar] [CrossRef]
- Wang, J.; Leung, D.; Chow, W.; Chang, J.; Wong, J.W. Development and Validation of a Multiclass Method for Analysis of Veterinary Drug Residues in Milk Using Ultrahigh Performance Liquid Chromatography Electrospray Ionization Quadrupole Orbitrap Mass Spectrometry. J. Agric. Food Chem. 2015, 63, 9175–9187. [Google Scholar] [CrossRef]
- Desmarchelier, A.; Fan, K.; Minh Tien, M.; Savoy, M.-C.; Tarres, A.; Fuger, D.; Goyon, A.; Bessaire, T.; Mottier, P. Determination of 105 antibiotic, anti-inflammatory, antiparasitic agents and tranquilizers by LC-MS/MS based on an acidic QuEChERS-like extraction. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2018, 35, 646–660. [Google Scholar] [CrossRef]
- Khaled, A.; Singh, V.; Pawliszyn, J. Comparison of Solid-Phase Microextraction to Solvent Extraction and QuEChERS for Quantitative Analysis of Veterinary Drug Residues in Chicken and Beef Matrices. J. Agric. Food Chem. 2019, 67, 12663–12669. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Rúbies, A.; Companyó, R.; Centrich, F. Determination of aminoglycoside residues in kidney and honey samples by hydrophilic interaction chromatography-tandem mass spectrometry. J. Sep. Sci. 2012, 35, 2710–2717. [Google Scholar] [CrossRef] [PubMed]
- Pugajeva, I.; Ikkere, L.E.; Judjallo, E.; Bartkevics, V. Determination of residues and metabolites of more than 140 pharmacologically active substances in meat by liquid chromatography coupled to high resolution Orbitrap mass spectrometry. J. Pharm. Biomed. Anal. 2019, 166, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Turnipseed, S.B.; Storey, J.M.; Wu, I.L.; Gieseker, C.M.; Hasbrouck, N.R.; Crosby, T.C.; Andersen, W.C.; Lanier, S.; Casey, C.R.; Burger, R.; et al. Application and evaluation of a high-resolution mass spectrometry screening method for veterinary drug residues in incurred fish and imported aquaculture samples. Anal. Bioanal. Chem. 2018, 410, 5529–5544. [Google Scholar] [CrossRef]
- Savoy, M.-C.; Woo, P.M.; Ulrich, P.; Tarres, A.; Mottier, P.; Desmarchelier, A. Determination of 14 aminoglycosides by LC-MS/MS using molecularly imprinted polymer solid phase extraction for clean-up. Food Addit. Contam. Part A 2018, 35, 675–686. [Google Scholar] [CrossRef]
- Desmarchelier, A.; Bessaire, T.; Savoy, M.-C.; Tarres, A.; Mujahid, C.; Beck, A.; Mottier, P.; Delatour, T. Screening of 154 Veterinary Drug Residues in Foods of Animal Origin Using LC-MS/MS: First Action 2020.04. J. AOAC Int. 2021, 104, 650–681. [Google Scholar] [CrossRef]
- Wang, C.; Li, X.; Yu, F.; Wang, Y.; Ye, D.; Hu, X.; Zhou, L.; Du, J.; Xia, X. Multi-class analysis of veterinary drugs in eggs using dispersive-solid phase extraction and ultra-high performance liquid chromatography-tandem mass spectrometry. Food Chem. 2021, 334, 127598. [Google Scholar] [CrossRef]
- Kong, C.; Wang, Y.; Huang, Y.; Yu, H. Multiclass screening of >200 pharmaceutical and other residues in aquatic foods by ultrahigh-performance liquid chromatography-quadrupole-Orbitrap mass spectrometry. Anal. Bioanal. Chem. 2018, 410, 5545–5553. [Google Scholar] [CrossRef]
- Turnipseed, S.B.; Lohne, J.J.; Storey, J.M.; Andersen, W.C.; Young, S.L.; Carr, J.R.; Madson, M.R. Challenges in implementing a screening method for veterinary drugs in milk using liquid chromatography quadrupole time-of-flight mass spectrometry. J. Agric. Food Chem. 2014, 62, 3660–3674. [Google Scholar] [CrossRef]
- Moretti, S.; Dusi, G.; Giusepponi, D.; Pellicciotti, S.; Rossi, R.; Saluti, G.; Cruciani, G.; Galarini, R. Screening and confirmatory method for multiclass determination of 62 antibiotics in meat. J Chromatogr A 2016, 1429, 175–188. [Google Scholar] [CrossRef]
- Pastor-Belda, M.; Campillo, N.; Arroyo-Manzanares, N.; Hernández-Córdoba, M.; Viñas, P. Determination of amphenicol antibiotics and their glucuronide metabolites in urine samples using liquid chromatography with quadrupole time-of-flight mass spectrometry. J. Chromatogr. B 2020, 1146, 122122. [Google Scholar] [CrossRef] [PubMed]
- Bortolotte, A.R.; Daniel, D.; Reyes, F.G.R. Occurrence of antimicrobial residues in tilapia (Oreochromis niloticus) fillets produced in Brazil and available at the retail market. Food Res. Int. 2021, 140, 109865. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Luiz, M.M.; Romero-González, R.; Plaza-Bolaños, P.; Vidal, J.L.M.; Garrido Frenich, A. Rapid and semiautomated method for the analysis of veterinary drug residues in honey based on turbulent-flow liquid chromatography coupled to ultrahigh-performance liquid chromatography-Orbitrap mass spectrometry (TFC-UHPLC-Orbitrap-MS). J. Agric. Food Chem. 2013, 61, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Luiz, M.M.; Romero-González, R.; Plaza-Bolaños, P.; Martínez Vidal, J.L.; Garrido Frenich, A. Wide-scope analysis of veterinary drug and pesticide residues in animal feed by liquid chromatography coupled to quadrupole-time-of-flight mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 6543–6553. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A. High-resolution mass spectrometry for bioanalytical applications: Is this the new gold standard? J. Mass Spectrom. 2020, 55, e4533. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Jianan, N.; Dapeng, P.; Ting, C.; Ijaz, A.; Aashaq, A.; Zhixin, L.; Muhammad Abu bakr, S.; Guyue, C.; Zonghui, Y. Current advances in immunoassays for the detection of antibiotics residues: A review. Food Agric. Immunol. 2020, 31, 268–290. [Google Scholar]
- Majdinasab, M.; Mishra, R.K.; Tang, X.; Marty, J.L. Detection of antibiotics in food: New achievements in the development of biosensors. TrAC Trends Anal. Chem. 2020, 127, 115883. [Google Scholar] [CrossRef]
- Yue, F.; Li, F.; Kong, Q.; Guo, Y.; Sun, X. Recent advances in aptamer-based sensors for aminoglycoside antibiotics detection and their applications. Sci. Total Environ. 2020, 762, 143129. [Google Scholar] [CrossRef]
- Tian, L.; Khalil, S.; Bayen, S. Effect of thermal treatments on the degradation of antibiotic residues in food. Crit. Rev. Food Sci. Nutr. 2017, 57, 3760–3770. [Google Scholar] [CrossRef]
- Canton, L.; Alvarez, L.; Canton, C.; Ceballos, L.; Farias, C.; Lanusse, C.; Moreno, L. Effect of cooking on the stability of veterinary drug residues in chicken eggs. Food Addit. Contam.-Part A Chem. Anal. Control Expo. Risk Assess 2019, 36, 1055–1067. [Google Scholar] [CrossRef]
- O’Brien, J.J.; Campbell, N.; Conaghan, T. Effect of cooking and cold storage on biologically active antibiotic residues in meat. Epidemiol. Infect. 1981, 87, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Sobral, M.M.C.; Cunha, S.C.; Faria, M.A.; Ferreira, I.M.P.L.V.O. Domestic Cooking of Muscle Foods: Impact on Composition of Nutrients and Contaminants. Compr. Rev. Food Sci. Food Saf. 2018, 17, 309–333. [Google Scholar] [CrossRef] [Green Version]
- Hussein, M.A.; Morshedy, A.M.; Ahmed, M.M. Effect of cooking methods on some antibiotic residues in chicken meat. Jpn. J. Vet. Res. 2016, 64, S225–S231. [Google Scholar]
- Shaltout, F. Impacts Of Different Types Of Cooking And Freezing On Antibiotic Residues In Chicken Meat. Food Sci. Nutr. 2019, 5. [Google Scholar]
- Kleechaya, W. Pharmacokinetics of oxytetracycline in black tiger shrimp, Penaeus monodon, and the effect of cooking on the residues. Aquac. Res. 2005, 254, 24–31. [Google Scholar]
- Salaramoli, J.; Heshmati, A.; Kamkar, A.; Hassan, J. Effect of cooking procedures on tylosin residues in chicken meatball. J. für Verbrauch. Und. Leb. 2015, 11, 53–60. [Google Scholar] [CrossRef]
- Franje, C.A.; Chang, S.-K.; Shyu, C.-L.; Davis, J.L.; Lee, Y.-W.; Lee, R.-J.; Chang, C.-C.; Chou, C.-C. Differential heat stability of amphenicols characterized by structural degradation, mass spectrometry and antimicrobial activity. J. Pharm. Biomed. Anal. 2010, 53, 869–877. [Google Scholar] [CrossRef]
- Clarke, A.; Means, W.; Schmidt, G. Effects of Storage Time, Sodium Chloride and Sodium Tripolyphosphate on Yield and Microstructure of Comminuted Beef. J. Food Sci. 2006, 52, 854–856. [Google Scholar] [CrossRef]
- Botsoglou, N.A.; Fletouris, D.J. Drug Residues in Foods: Pharmacology, Food Safety, and Analysis; Marcel Dekker: New York, NY, USA, 2001; ISBN 0-367-39792-7. [Google Scholar]
- Uno, K.; Aoki, T.; Kleechaya, W.; Tanasomwang, V.; Ruangpan, L. Pharmacokinetics of oxolinic acid in black tiger shrimp, Penaeus monodon Fabricius, and the effect of cooking on residues. Aquac. Res. 2006, 37, 826–833. [Google Scholar] [CrossRef]
- Hasanen, F.; Mohammed, M.M.; Hassan, W.M.; Amro, F. Ciprofloxacin residues in chicken and turkey carcasses. Benha Vet. Med. J. 2016, 31, 136–143. [Google Scholar] [CrossRef]
- Roca, M.; Castillo, M.; Marti, P.; Althaus, R.L.; Molina, M.P. Effect of Heating on the Stability of Quinolones in Milk. J. Agric. Food Chem. 2010, 58, 5427–5431. [Google Scholar] [CrossRef]
- Ismail-Fitry, M.R.; Jinap, S.; Bakar, J.; Saleha, A. Effect of Deep-Frying at Different Temperature and Time on Sulfonamide Residues in Chicken Meat-Balls. J. Food Drug Anal. 2008, 16, 81–86. [Google Scholar] [CrossRef]
- Javadi, A.; Mirzaie, H.; Khatibi, A. Effect of roasting, boiling and microwaving cooking method on sulfadiazine+ trimethoprim residues in edible tissues of broiler by microbial inhibition method. African J. Microbiol. Res. 2011, 5, 16–19. [Google Scholar]
- Zhao, X.H.; Wu, P.; Zhang, Y.H. Degradation kinetics of six sulfonamides in hen eggs under simulated cooking temperatures. J. Serbian Chem. Soc. 2011, 76, 1093–1101. [Google Scholar] [CrossRef]
- Roca, M.; Althaus, R.; Molina, M.P. Thermodynamic analysis of the thermal stability of sulphonamides in milk using liquid chromatography tandem mass spectrometry detection. Food Chem. 2013, 136, 376–383. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Class | Substance | Aquaculture | Bovine | Eggs | Honey | Milk | Pig | Poultry | Rabbits | Sheep/goats | Eggs |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sulfonamides | Sulfadimethoxine | √ | √ | √ | √ | √ | √ | ||||
| Sulfadimidine | √ | √ | |||||||||
| Sulfamethoxypyridazine | √ | ||||||||||
| Sulfacetamide | √ | ||||||||||
| Sulfachlorpyrazine | √ | ||||||||||
| Sulfadiazine | √ | √ | √ | √ | √ | ||||||
| Sulfadimidine | √ | ||||||||||
| Sulfamethazin | √ | ||||||||||
| Sulfathiazole | √ | ||||||||||
| Tetracyclines | Oxytetracycline | √ | √ | √ | √ | √ | √ | √ | |||
| Epi-Oxytetracycline | √ | ||||||||||
| Chlortetracyclin | √ | ||||||||||
| Doxycycline | √ | √ | √ | √ | |||||||
| Penicilines | Amoxycillin | √ | √ | √ | |||||||
| Benzylpenicillin (Penicillin G) | √ | √ | |||||||||
| Ampicillin | √ | ||||||||||
| Cloxacillin | √ | ||||||||||
| Macrolides | Gamithromycin | √ | |||||||||
| Spiramycine | √ | ||||||||||
| Neospiramycin | |||||||||||
| Tilmicosine | √ | √ | √ | √ | √ | ||||||
| Tulathromycin | √ | √ | |||||||||
| Erythromycin | √ | ||||||||||
| Quinolonas, including fluoroquinolones | Enrofloxacin | √ | √ | √ | √ | √ | √ | √ | |||
| Oxolinic Acid | √ | ||||||||||
| Ciprofloxacin | √ | √ | |||||||||
| Oxolinic Acid | √ | ||||||||||
| Danofloxacin | √ | √ | |||||||||
| Sarafloxacin | √ | ||||||||||
| Flumequine | √ | √ | |||||||||
| Aminoglycodides | Dihydrostreptomycin | √ | √ | √ | √ | ||||||
| Neomycin | √ | ||||||||||
| Streptomycin | √ | ||||||||||
| Aminosidin (Paromycin, Paromomycin) | √ | ||||||||||
| Phenicols | Florfenicol | √ | |||||||||
| Lincosamides | Lincomycine | √ | |||||||||
| Diaminopyrimidinas | Trimethoprim | √ | √ | √ | √ |
| Type of Sample | Class of Antibiotic Residues Analyzed | Extraction Methods | Extraction Conditions | Clean-Up | # Samples | Sampling Period | Levels of Contamination | Recoveries | Conclusions of the Study | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Milk | BZM, AT | QuEChERS d-SPE | 10 g + 10 mL AcN 0.1% NH3 + 5 g MgSO4: NaCl (4:1, w/w). Shake for 1 min, centrifuge at 5000 rpm for 5 min. | Transfer 1 mL of the supernatant to a 2 mL eppendorf containing MgSO4 (150 mg), C18 (50 mg) and PSA (50 mg). Vortex for 1 min and centrifuge at 3000 rpm for 2 min. Evaporate an aliquot of the supernatant (500 µL) to dryness and reconstitute in acetonitrile:water (5:95). | 2013 | 10–100 µg/Kg | 75–100% | The application of a modified version of the QuEChERS method assured a fast sample treatment with low MLOQ (<3 µg/Kg), allowing the determination of these compounds at concentration levels considerably lower than the established EU MRL values. | [70] | |
| Poultry muscle | MA | SPE | 5 g + 20 mL EDTA-McIlvaine’s buffer pH 3.5. Mix and centrifuge at 5000 rpm for 10 min. Centrifuge again twice more with 20 + 10 mL EDTA McIlvaine’s buffer, respectively. | Conditioning the Oasis HLB cartridges (500 mg) with 5 mL of MeOH and 5 mL of water. Pass the extract. Wash the columns with 5 mL of water and elute with 5 mL of MeOH. Evaporate the extract to dryness. Re-dissolve with 1 mL of the 0.05% TFA (pH 2.5)/ACN (70:30, v/v) mixture. | 30 | 2013 | 200 µg/Kg | 85–120% | The spiramycin and tylosin were not detected in the analyzed samples. Only tilmicosin was found in two broiler chickens at 38 µg/Kg (supermarket) and 65 µg/Kg (wet market) with RSDs (%) (n = 2) of 0.2% and 0.4%, respectively. The tilmicosin did not exceed the maximum permitted limit (MRL) of drug residues in chicken muscle, which was 150 µg/Kg according to EU Regulation and 100 µg/Kg according to the Malaysian Food Act. | [60] |
| Milk and muscle | MA, LS | LLE | 2 mL of milk + 4.0 mL of ACN, divided into 3 aliquots (2.0 mL + 1.0 mL+ 1.0 mL). Shake for 20 min and centrifuge for 5 min at approximately 3000 g, at 5 °C. 2 g of muscle + 10.0 mL of ACN. Homogenize with a mechanical mixer. Shake for 20 min and centrifuge for 5 min, at approximately 3000 g, at 5 °C. | 2015 | 50–100 µg/Kg | 60–73% (Milk) 60–107% (Muscle) | After LLE, followed by centrifugation, the obtained organic extract was very limpid and adequate to be directly injected into the LC–MS/MS system. The sample preparation method has shown high efficiency in extracting the target compounds. Fast and environmentallyfriendly once it generates fewer solvent residues than other methods that use SPE. | [31] | ||
| Milk | PN | LLE | 2.0 mL + ACN (2 mL + 1 mL + 1 mL) with manual mixing between each addition. Mix 15 min. Centrifuge 5 min at 4000 g, at low temperature (5 °C). | Transfer the supernatant to a 50 mL tube with 100 mg of C18 bulk. Shake and centrifuge for 5 min at 4000 g. Take the supernatant in a freezer (−17 °C) for 20 min. Centrifuge (10 min at 4000 g, 0 °C). Transfer the supernatant and evaporate to approximately 500 µL | 247 | 2015 | 0.10, 0.25xMRL | 58–108% | From a total of 247 samples, two samples had positive residues; one sample contained cloxacillin (107 ng/L), and another contained cefapirin. The sample preparation method was not complex and is environmentally friendly when compared with reports for β-lactam analysis in food samples, which generally require the use of SPE associated with additional clean-up steps. Validation data were satisfactory, showing agreement with the criteria proposed by Commission Decision 2002/675/EC. | [57] |
| Butter, egg and milk powder | QN, TC, CP, MA, SA, BZM, NMZ, CCD, NSAIDs, AM, CDC, Antiepileptic drugs, Fibrates and Others (115) | LLE | 1 g + 2 mL of H2O containing 0.1% formic acid (v/v) and 0.1% EDTA (w/v). Mix and add 2 mL ACN + 2 mL MeOH. Mix for 30 s. Put the samples in an ultrasonic bath at 600 C for 20 min. Centrifuge at 4000 rpm for 10 min. | Transfer the supernatant and precipitate at 23 °C for 12 h. Centrifuge again. Defatting with 5 mL of hexane. Vortex for 1 min. Centrifuge at 4000 rpm for 10 min. Discard the n-hexane layer. Evaporate to dryness. Re-dissolution in 1 mL of MeOH/aqueous formic acid solution, 0.05% (25:75 v/v) | 108 | 1–200 μg/Kg | Butter: 50–120% Fish tissues: 50–9% Eggs: 50–80% (66 compounds) Milk: 50–80% (78 compounds) | Oxfendazole and albendazole sulfone were detected in two milk powder samples at 1 μg/Kg and 0.54 μg/Kg, respectively. These concentrations, although they are higher than LOQ, are very low and far below the MRL established for these benzimidazoles in milk (10 μg/Kg and 100 μg/Kg). Several quinolones were detected in milk (ciprofloxacin at 7.3 μg/Kg and norfloxacin at 2.2 μg/Kg) and fish tissue samples (flumequine at 4.6 mg/kg and enrofloxacin at 4.8 mg/kg). Thiabendazole was detected and quantified at 9.7 mg/kg in one butter sample, and, finally, caffeine was present in six milk powder samples at concentrations ranging from 2.4 to 32 mg/kg. No compound detected and quantitated would exceed the MRL established in Commission Regulation 37/2010/EC. | [47] | |
| Eggs | Amoxicillin and metabolites | LLE | 5 g + ACN. Mix for 15 min. Centrifuge at 3500 g at 4 °C for 10 min. Repeat the extraction. Adjust the volume to 1 mL with ammonium acetate (3.89 mol/L, pH 6.74). Add 20 mL of dichloromethane. Shake and centrifuge at 3500 g, 4 °C for 5 min. Transfer 1 mL of the supernatant and evaporate to dryness. Reconstitute with 2 mL of 2% acetonitrile in water. | 40 | 2015 | 0.1–0.6 µg/Kg | 80.15–98.23% | Compared with previous publications, this method presented fewer interferences and higher recovery rates (≥80%). The major advantage of this protocol is its simplicity and short analysis time. One sample was contaminated with 4.53 µg/Kg amoxicillin (<MRL), while amoxicilloic acid, diketopiperazine-20,50-dione and ampicillin were not found in any samples. | [58] | |
| Muscle, liver and kidney | AG | SLE, SPE | 5.0 g + 10 mL of 5% TCA (w/v). Mix and centrifuge at 5 °C, 8000 rpm. Repeat with 10 mL of 5% TCA. Add 5 mL of 0.2 mol/L HFBA + 5 mL n-hexane. Shake, and centrifuge at 3600 rpm for 30 min. Remove the n-hexane phase. | Conditioning the HLB columns with 3 mL of MeOH + 3 mL of water + 3 mL of 0.2 mol/L HFBA. Transfer 5 mL of the extract. Adjust the collected eluent to pH 8.5 ± 0.2 with 100 g/L NaOH + 0.2 mol/L HCl. Dry the columns. Condition other HLBs with 3 mL of MeOH + 3 mL of water + 3 mL of 0.2 mol/L of HFBA + 3 mL of aqueous NaOH pH 8.5. Transfer the effluent to pH 8.5 ± 0.2 in the column. Connect the two HLB cartridges with vacuum joints. Wash the two tandem cartridges with 5 mL of water and dry. Elute with 6 mL ACN/0.15 mol/L HFBA (4: 1, v/v). Evaporate to 0.3 mL. Reconstitute with 1 mL 20 mmol/L HFBA. | 2016 | 0.5, 1, 1.5 x MRL | Muscle: 47–64/7–85% Liver: 5–68/70–85% Kidney: 56–70/70–85% | The method showed good sensitivity, and the performance characteristics comply with EU recommendations. This method should be an efficient approach for multiresidue analysis of AGs in animal tissues. | [71] | |
| Bovine muscle | AG, PN, MA, SA, AT, CD, NSAIDs, Pharmaceuticals, Rifaximin, Baquiloprim, Trimethoprim, Chlorpromazine, 6-phenyl-2-thiouracil, Bromhexine (76) | SLE, SPE | 5 g + 10 mL of ACN. Centrifuge. Decant the supernatant. Evaporate to 1.0 mL. Add 20 mL of the 0.4 mM EDTA solution + 1% NaCl (w/v) + 2% TCA (w/v). Shake for 60 min on a mechanical stirrer. Centrifuge at 4000 rpm for 5 min. Decant the supernatant, and adjust the extract to pH 6.5 by adding approximately 6 drops of 30% NaOH (w/v). | Conditioning OASIS HLB Cartridge (200 mg, 6 mL), with 6 mL MeOH + 6 mL H2O. Pass the sample through the columns and dry it in a vacuum for 15 min. Elute with 2 × 0.5 mL of 10% (v/v) aqueous formic acid + 3 × 1 mL of ACN. The eluate is collected and combined with 1 mL ACN extract. | 10 | 2016 | 5–1000 µg/kg | 37.4% (bromhexine) to 106% (kanamycin). | No positive results were found in any of the analyzed samples. The low recoveries of some compounds are considered acceptable since they were reproducible. | [72] |
| Animal tissue (muscle), honey, milk | AG | LLE, SPE | Honey: 2 g + 10 mL of solution A (50 mmol/L potassium phosphate). Shake for 5 min. Adjust the pH = 7.0 with ammonia solution (28.0–30.0 %). Animal tissue and milk: 2 g + 5 mL of solution B (10 mmol/L potassium phosphate monobasic (KH2PO4) + 0.4 mmol/L EDTA-Na2 + 2% TCA). Vortex for 5 min. Centrifuge at 3200 rpm, 4 °C, 5 min. Transfer the supernatant, add 5 mL of solution B and repeat the extraction process. Dilute the supernatant with 1:1 solution A. Adjust the pH = 7.0 with ammonia solution (28.0–30.0%). | Conditioning SupelMIP SPE-AGs cartridges (3 mL/50 mg) with 1 mL MeOH + 1 mL 50 mM K3PO4 (pH 7). Pass the extract. Wash the columns with 3 mL of water + 1 mL of 0.1% ammonia solution at 0.5 mL/min (the ammonia washing step is not carried out for milk and pork samples). Apply strong vacuum, 5 min. After washing step with 0.1% ammonia. Wash with 1 mL of acetonitrile-water (40 + 60, v/v) + 1 mL of methanol-dichloromethane (50 + 50, v/v). Elute with 1 mL of 1% formic acid in MeOH-water (80 + 20, v/v). | 2016 | 20–300 µg/kg | 78.2–94.8% | The MIP proved to be a powerful and selective material for the cleanup of extract from complicated food matrixes. | [73] | |
| Muscle, urine | PN, CP, SA, TC, MA, LS, QN, AM, DP (29) | SPE | Muscle: 1 g + 5 mL McIlvaine buffer (pH 4.0) + TCA (100 mL, 20% w/v). Vortex. Centrifuge at 2500 g, 4 °C, 10 min. Transfer the supernatant to a new centrifuge tube. Degrease with 2 × 3mL of n-hexane. Discard the n-hexane layer at each centrifugation (2500g, 5 min). Urine: 5 mL centrifugue at 2500g, 4 °C to 5 min. | Muscle: Conditioning the Oasis HLB cartridges with 3 mL of MeOH + 3 mL of water under vacuum. Transfer the extract to the cartridge. Wash the cartridge with 2–3 mL of MeOH: water (5:95 v/v). Elute with 5 mL of MeOH and collect. Evaporate the eluate. Reconstitute in 200 mL of MeOH: water (10:90 v/v). Urine: Conditioning the Oasis HLB cartridges (3 mL, 60 mg) with 3 mL of MeOH + 3 mL of 0.5 M HCl + 3 mL water, under vacuum. Transfer 5 mL of urine to the cartridges. Wash with 3 mL of water + 3 mL of MEOH: water (20:80, v/v). Elute with 5 mL of MEOH and collect. Evaporate the eluate. Reconstitute in 200 mL of MeOH: water (10:90 v/v). | 86 (43 urine; 43 muscle) | 2017 | Muscle: 1–10 ng/mL Urine: 0.5 ng/mL | Muscle and urine: 90–106% | Doxycycline was one of the antibiotics most frequently found in urine (in 37 samples, at a maximum of 339.45 µg m/L). In the muscle tissues, doxycycline was found 15 times, and the maximum recorded was 21.05 µg/kg. No strict correlation was found between the two matrices, but generally, when the concentration in the urine was very high, the analyte was detected in the paired muscle sample. | [44] |
| Meat, Milk, Egg, Fish | AM, AT, BZM, PN, CCD, IO, MA, NAIDS’s, QN, SA, TC, TQ (100) | d-SPE, LLE | 2 g tissue (muscle, liver, or kidney) + 10 mL mixture of ACN and water (4:1, v/v) containing 2 mM ammonium formate. 1 g of milk + 2 mL ACN the resulting mixture, was put into an ultra-centrifugal filter (cut-off membrane at 3 kDa, 4 mL) and centrifuged. 2 g of egg and fish + 3 mL of ACN + 1 mL of water contains 2 mM ammonium formate. Centrifuge 5000× g at 4 °C, 5 min. The supernatant (2 mL) was purified by centrifugation (5000× g, 4 °C, 60 min) in an ultra-centrifugal filter (cut-off membrane at 3 kDa) + 2 mL of hexane. Vortex shake and re-centrifuged. | The tissue in solvent mixture was homogenized, shaken for 5 min, and centrifuged. 10 mL hexane + 250 mg octadecyl carbon chain (C18)-bonded silica.Vortex shake for 30 s. The mixture was centrifuged for 5 min at 10,000 rpm. Discharged the n-hexane. The extract was evaporated under nitrogen until near dryness. | 2016 | 5, 10, 20, 50, 100 µg/kg | 63 to 122% | The simple pretreatment and rapid detection method significantly reduced the time (2–3 h), human resources, and hazardous organic solvent requirements compared with conventional methods. Good ccα’s and good recoveries in general for the three matrices. | [74] | |
| Meat (pork, chicken, fish tissues) and Eggs | SA, QN | UAE, SPE | 2 g + 10 mL of 0.1% formic acid in 90:10 ACN/water (v/v). The mixture was subjected to UAE for 10 min at 20 °C and then centrifuged at 8000 rpm for 15 min at 4 °C. Remove the supernatant. | Pass a 2 mL aliquot of the supernatant in Oasis PRMiME HLB 3 cc Vac Cartridge, 60 mg. Collect the eluate and evaporate to dryness under mild ultra-high purity nitrogen gas at 35 ° C. The resulting residues were reconstituted in 1 mL of initial mobile phase (10 % MeOH). | 151 | 2017 | 2.5, 5.0 and 10.0 μg/kg | Pork: 61.9–85.1%/95.4–121.6% Chicken: 51.9–58.6%/95.4–121.6% Fish: 42.2–51.1%/106.7–116.1% Eggs: 57.1–73.9%/108.7–122.4% | Sulfacetamide at 1.2 μg/kg in one pork sample, sulfaquinoxaline at 3.3 μg/kg in another pork sample, and sarafloxacin at 0.98 μg/kg in one egg were detected. These concentrations are very low and far below the MRL. | [68] |
| Fish muscle | TC (5) | QuEChERS | 1 g + 2.2 mL of EDTA-McIlvaine buffer + 5.0 mL for ACN + 1.25 g of (NH4)2SO4. Shake for 1 min, centrifuge at 9000 rpm for 5 min. | Transfer 5.5 mL of the supernatant to polypropylene tubes with 50 mg of C18. Vortex for 1 min, centrifuge for 5 min at 5000 rpm. Transfer 5 mL of the supernatant and bring to dryness. Reconstitute with 500 µL of H2O: MeOH (95: 5, v/v) | 10 | 2018 | 5, 20, 100 μg/kg | 80–105% | Only traces of oxitetracycline were found in one salmon sample, obtaining a concentration of 5.6 ± 2.2 µg/kg. LOD between 0.6–1.3 μg/kg and LOQ between 1.7–4.0 μg/kg. Good precision. | [42] |
| Muscle, kidney, liver | TU, NMZ, LS, AT, TC, PN, QN, BA, CP, SA, MA, AM, CDC, TQ, Flukicide, NSAID, Progestin | SPE | 2 g + 2 mL 0.1 M EDTA. Shake and centrifugate. Transfer the supernatant. Add 8 mL of cold ACN w/2% FA and 2% DMSO to the sample residue left in the first tube. Shake and centrifuge. Decant supernatant was placed in the second tube and combined with the previous extract. Vortex the mixed extract in the second tube and centrifuge. | Transfer 5 mL of supernatant to EMR-Lipid 6 mL cartridge. Add 1.25 mL 20:80 water/ACN into EMR-Lipid 6 mL cartridge. Collected eluent and combined 0.5 mL of sample eluent with 0.3 mL of water. | 2018 | 20/4 ng/g (G1/G2) | 60–120% (for 90% of tested veterinary drugs) | EMR-Lipid cartridge cleanup provides high matrix co-extractive removal and reduces the matrix ion suppression on the target analytes. The quantitative analysis results showed that >90% of tested veterinary drugs provided acceptable recoveries, and >95% of compounds gave excellent reproducibility in all five meat matrices. | [51] | |
| Poultry muscle | TT, BA, QNX, PN, LS, MA, NMZ, BZM, NSAIDs, QN, SA, TC, CCD, AT, CDC, PP, Pesticides and others (140) | SLE, d-SPE | 5g + 0.2 mol/L ethylenediamine tetraacetic disodium salt (EDTA–Na2) + 12.5 mL of ACN:ethanol (80:20, v/v). Defatting twice with 10.0 mL of n-hexane and centrifuge. Transfer ACN–ethanol aqueous phase to another tube with another 10 mL of ACN:ethanol (80:20, v/v). Remove the supernatant, transfer it to another tube, and centrifuge. | Remove 7.5 mL of the liquid to another tube with 750 mg primary–secondary amine (PSA): aminopropyl (NH2) (50:50, m/m). Shake and centrifuge for 5 min at 0 °C. Transfer 6 mL of the supernatant and evaporate to below 1.1 mL. Add 0.4 mL of DMSO: MeOH (25:75, v/v) to the tube concentrate to 1.5 mL with water and shake for 20 s. | 50 | 2017 | 4.8–16 µg/kg | 60–139% | Doxycycline, tetracycline, ofloxacin, enrofloxacin, sulfaclozine, sulfaquinoxaline, and amantadine were found in 22 chicken samples. Two positive samples were found in chicken muscle containing sulfaclozine and sulfaquinoxaline at 4524 and 286 μg/kg, respectively, which exceeded the EU Maximum Residue Limits (MRL) (100 μg/kg). Amantadine was found in five chicken samples (58–927 μg/kg). Other drugs were lower than their corresponding EU MRL. The very hydrophobic analytes were not included in this study. | [75] |
| Nile tilapia | AM (CAP, FF, TAP) | SLE | 1 g + 10 mL of ethyl acetate: ammonium hydroxide (98:2). Vortex, centrifuge 7000 g, 4 °C, 1 min. Add 500 µL of 5% acetic acid to the supernatants. Shake and evaporate to 1–2 mL at 55 °C. Add 250 µL of 5% acetic acid to the concentrated extract, vortex 30 s. Add heptane (5 mL), vortex 1 min, centrifuge at 7000g at 4 °C for 1 min. Discard the heptane phase. Evaporate to dryness. | 32 | 2018 | 0.60–75 µg/kg | 79.8–92.0% | One (3.1%) sample was positive for thiamphenicol <LOQ (12.5 μg/kg). All samples were positive for florfenicol at levels below the LOQ (12.5 μg.kg−1). The presence of florfenicol in every sample analyzed, even below MRL, suggests this drug has been used widely in tilapia production, not only in fish farming. | [76] | |
| Muscle, kidney, liver | QN, SA, TC, MA, LS, CP, PN, AM, AG, TU, BA, AT, CD, NMZ, TQ, Anti-inflammatories and others (174) | SLE, SPE, d-SPE | Aminoglycoside: 2 g + 20 mL of 10 mM NH4OAc, 0.4 mM EDTA, 0.5% NaCl, and 2% TCA. Vortex for 5 min and centrifuge for 3 min at 4150 rpm. Transfer 10.75 mL (1 g of equivalent sample) of extract, adjust the pH to 6.5 ± 0.1 with a few drops of 30% aqueous NaOH solution. Centrifuge again. MMM (Multiclass, Multiresidues): 2 g + 10 mL of 4/1 (v/v) ACN/water. Vortex for 5 min and centrifuge for 3 min at 4150 rpm. | Aminoglycoside: Conditioning each WCX (50 mg) (Weak Cation Exchange) tip on the DPX (Dispersive Pipette Extraction) device with a few pumps of 3 mL MeOH + 3 mL of water sequentially. Repeatedly pull the 10.75 mL extracts to the DPX tips in four portions of ± 2.7 mL each. Wash the sorbents at the tips with 3 mL of water and dry. Aspirate 1 mL of 10% aqueous formic acid solution into and out of the tips five times. Eluate DPX tips with 1 mL of 10 % FA in water. Combine 407 μL of the MMM extract (0.172 g/mL of the equivalent sample) and 70 μL of the aminoglycoside extract (1 g/mL) in a 1 mL autosampler vial. Add 273 μL of 146.5 mM 1-heptanesulfonate aqueous reagent IP solution (ion-pairing reagent). | 2017 | 0.5, 1, and 2 times the regulatory levels of interest (10–1000 ng/g, depending on the drug) | 70–120% in 79–84% of the analyzed analytes | Bad results for aminoglycosides in muscle because the muscle extracts were very fatty and blocked the ends of the DPX during sample preparation. In the case of bovine muscle, 100/131 medications (76%) previously met the criteria for quantitative validation, and in this study, 137–149 of 174 medications (79–84% depending on the matrix) met the criteria. | [77] | |
| Chicken Tissues | MA | SPE | 2.0 g + 200 μL of 0.1 mol/L EDTA solution. Shake 20 min, centrifuge at 5000 r/min for 5 min. Extract the supernatant twice with 10mL of ACN/MeOH (v/v, 95:5). Evaporate to 1 mL after adding 0.4 g of NaCl. Wash with 1mL of acetonitrile and 15mL of water and collect the eluent. | The pretreatment procedure of PAF-6 SPE cartridge (60 mg/3 cc): first, the cartridges were prepared by packing 60 mg of PAF-6 into the empty polypropylene SPE cartridges (3 mL). Pass 8 mL of the extract through the cartridges, preconditioned with 3 mL of methanol + 3 mL of water. Wash with 5 mL. Elute with 5mL of 5% methanol ammonia. Evaporate and redissolve to 1.0 mL with the mobile phase. | 6 | 2018 | 1–50 µg/kg | 82–101.4% | Tylosin was detected in two samples with contents of 38.752 μg/kg and 79.211 μg/kg, respectively. Azithromycin and tilmicosin were detected in one sample; the contents were 27.336 μg/·kg and 56.719 μg/kg, respectively. | [61] |
| Pangasius fillet | PN, QN, MA, SA, TC, AM, trimethoprim | SLE | 2.0 g + 0.5 mL 0.1 M EDTA. Vortex shake for 2 min. Add 3.5 mL of ACN, vortex; shake for 5 min. Centrifuged for 5 min at 13,000× g at 18 °C. | 40 | 2019 | Low: 3 ng/g Low middle: 10 ng/g High-middle: 50 ng/g High: 100 ng/g | Low: 80.7–119.8 % Low-middle: 78.8–118.3 % High-middle: 76.9–114.2 % | Samples showed enrofloxacin residue levels, all of which were above the LOD of the method (1.00 ng/g) but below the LOQ (3.00 ng/g). The analysis of pangasius samples imported from Vietnam and acquired in the Brazilian retail market indicated the presence of low residue levels of enrofloxacin in five out of 40 samples analyzed. Considering the FDA, Brazilian and Vietnamese regulatory framework, and the fact that there is no safe level (MRL) set by Codex Alimentarius for enrofloxacin residues in fish that may pose an acceptable risk for consumers, those contaminated samples must be considered out of conformity. | [37] | |
| Muscle, Eggs | QN | SLE, SPE | 5.0 g + 20 mL EDTA Mcllvaine buffer (0.1 mol/L, pH 4.0). Vortex shake. Add 10 mL of MeOH, centrifuged at 8000 rpm for 10 min at 4 °C. Filter the supernatant on a qualitative quick filter paper. Dilute the filtrate with 100 mL with water. | Conditioning Oasis HLB cartridges with 3 mL of MeOH + 3 mL of water. Pass 20 mL of the extract. Wash with 1 mL of 5% (v/v) MeOH in water, elute with 6 mL of MeOH. Evaporate and redissolve in 1.0 mL of 0.2% formic solution. | 170 | 2019 | 2.0, 5.0 µg/kg | Muscle: 70.4–98.4% Egg: 66.9–99.0 % | Enrofloxacin was detected in six chicken meat samples, and its level varied from 4.88 to 44.4 µg/kg; traces of ciprofloxacin (lower than 7 µg/kg) were also found. In addition, eight of 110 egg samples contained traces of enrofloxacin at levels ranging from 1.09 to 5.22 µg/kg. Traces of ciprofloxacin were also present in egg samples (<7 μg/kg). | [64] |
| Honey | AG | SLE, d-SPE | 0.2 g of sample + 2mL H2O. Vortex shake and ultrasound. Add 2ml of ACN, vortex; shake for 2 min, and centrifuge 20 min at 15 °C. | Magnetic Fe3O4@SiN- galactitol nanoparticles for DSPE (1 mg): Pass the supernatant in the DSPE. Elute with 150 µL FA 190mM | 2019 | 15–60 µg/kg | 84–109% | Compared to other SPE methods previously reported for AGs analysis, the present one employed a minimum amount of sorbent (1 mg) and sample (0.2 g). The final optimized method was validated for the analysis of four aminoglycosides in honey with acceptable and reliable results. | [78] | |
| Duck meat | TT, SA, PP, QN, MA, LS, trimethoprim | SLE, SPE | 2 g + 6mL of 0.1 M Na2EDTA solution + 4mL of 2% TFA solution. Vortex shake for 5 min and centrifuged at 2600 g at 4 °C for 10 min. The obtained supernatant was transferred to a new tube containing 10 mL of n-hexane, vortexed for 5 min, and centrifuged at 2600 g for another 10 min. | Conditioning the Oasis HLB columns (6 cm3, 200 mg) with 3 mL ACN + 3 mL MeOH + 2 mL of distilled water. Pass 10 mL of the extract. Wash the columns with twice 5 mL of distilled water and dry for 10 min. Elute with 3mL MeOH + 3 mL ACN + 3 mL of 0.02% ammonia solution in MeOH. Evaporate to dryness and redissolve in 2 mL of MeOH. | 2019 | 5, 10, 20 µg/kg | 69.8–103.3% | Values of LOD ranging from 1.63 to 8.65 μg/kg and LOQs ranging from 4.93 to 26.21 μg/kg were achieved. LOQ values were much lower than the MRLs. The matrix effect varied between −47.2% to −13.5%, and an ion suppression was observed for all analytes in the duck meat matrix. | [50] | |
| Milk | BZM, CP, IZ, MA, NSAIDs, PN, QN, SA, BA, steroids (66) | d-SPE, SPE | Procedure I: 1 g + 4 mL ACN (2% FA). Mix in vortex. Centrifuge at 6460 g for 5 min. Procedure II: 2 g + 10 mL ACN (5% FA). Mix in vortex. Centrifuge at 6460 g for 5 min. | Procedure I: Conditioning the HLB PRiME columns (3 cc, Waters) with 3 mL of ACN (2% FA). Pass 4 mL of the organic layer. Dilute 100 μL of the eluted extract from the cartridge with 100 μL of ACN + 800 μL of H2O so that the final composition is 80:20 (v/v) aqueous: organic. Procedure II: Solvent extraction followed by EMR-Lipid dSPE cleaning (EMR-Lipid dSPE and EMR-Lipid Polish tubes, Agilent Technologies): Condition the EMR-Lipid dSPE sorbent with 5 mL of ammonium acetate buffer solution (5 mM). Add 5 mL of the organic layer to the EMR-Lipid dSPE tube. Shake manually for 1 min and vortex for 1 min. Centrifuge at 2650 g for 3 min. Add 5 mL of the top layer of the tube to the EMRLipid Polish tube (containing 1.6 g of MgSO4 + 0.4 g NaCl). Vortex for 2 min. Centrifuge at 2650 g for 3 min. Collect the organic phase. Make a final 1:10 dilution with a 100 μL aliquot of the extract, resulting in a final composition of 80:20 (H2O/ACN, v/v). | 24 | 2018 | 50 μg/kg | Procedure I- 72.4–115.9 % Procedure II: 75.6–119.2 % | Traces of danofloxacin were found in two whole cow milk, in the range of 0.7–1.5 μg/kg. However, these concentrations were below the current MRL established. Both methodologies provide satisfactory results in terms of matrix effect, sensitivity, recoveries, precision and being environmentally friendly. The HLB PRiME procedure is faster than EMR-Lipid. The tolerance to the matrix effect was higher with the SPE cleaning. | [79] |
| Meat (Pork) | AM, BA, BZM, CCD, CDC, IO, LS, MA, NMZ, QN, SA, TC, TQ, antivirus drugs, resorcylic acid lactones, steroid hormones, triphenylmethane dyes, and others | SLE, SPE | 2.0 g + 0.5 mL 0.1 M EDTA + ACN/water solution (6 mL, 80/20, v/v). Shake end-over-end for 10 min and centrifuge at 4600 rpm for 10 min. | Conditioning C18 SPE cartridges (Waters, Milford, MA, USA) with ACN/H2O (3 mL, 80/20, v/v). Pass the extract and collect. Evaporate to dryness and re-dissolve in 500 μL of water-ACN solution mixture (95/5, v/v) containing 5 mM ammonium formate and 0.1% formic acid. | 40 | 2020 | LLOQ (µg/kg) = 10, 100 or LLOQ (µg/kg) = 50, 100 | 70% for all of the compounds with the exception of triamcinolone, triamcinolone acetonide, fluocinolone acetonide and clobetasol propionate. | Sulfamethazine was detected in one sample, and its metabolites were successfully found in one run. Sulfamethazine content (1150 µg/kg) was much higher than the MRL established in pork (100 µg/kg). | [80] |
| Muscle, Fat, Liver and Kidney | PN (Amoxicillin) | SPE | 2.0 g + 8 mL phosphate buffer (0.01 M, pH = 6.3). Refrigerated centrifuge at 10,960 g for 10 min at 4 °C. Vortex for 2 min and refrigerated centrifuge at 10,960 g for 10 min at 4 °C after addition of 1 mL TCA solution (50 mg/mL). | Condition Oasis® HLB cartridges (60 mg, 3 mL) with 3 mL of MeOH + 3 mL of water. Pass the extracts. Wash the columns with 2 mL of water. Elute with 3 mL of ACN. Evaporate to dryness and re-dissolve with 2 mL of initial mobile phase. | Muscle, fat, liver, kidney: 10–100 µg/kg | Muscle (µg/kg): 91.06–100.81 Fat (µg/kg): 102.39–101.79 Liver (µg/kg): 113.94–111.72 Kidney (µg/kg): 98.67–89.49 | The results obtained from the present study revealed satisfactory recovery and precision, which were consistent with the EU requirements, indicating that the method using stable isotopically labeled analogs as an internal standard was reliable. | [81] |
| SPME | SBSE | MSPD | Micro-SPE | SPE | d-SPE | d-SPME | MSPE | MIP-SPE | LLE | QuEChERS | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sensitivity | high | higher (SPME) | high | ||||||||
| Automation | convenient | difficult | |||||||||
| Extraction Time/Sample preparation | short | long | short | short | short | extremely fast | fast | fast | |||
| Efficiency of Extraction | High control | may be affected by poor packing | high efficiency | high efficiency | high efficiency | ||||||
| Cost | low | low | low | low | low | low | |||||
| Coupling with chromatographic instruments | easy | easy | highly compatible (HPLC) | ||||||||
| Solvent consumption | low | low | low | possibility | minimal | minimal | large | ||||
| Simplicity | reduced labor-intensive manual operations | yes | yes | yes | easy to perform | easy to perform | easy to perform | ||||
| Single step | sampling and extraction | extraction and cleaning-up | extraction and concentration | extraction and cleaning-up | |||||||
| Sorbent choice | difficult | limited availability | broad variety | easy to perform | |||||||
| Robustness/Reproducibility | low | yes, to different types of samples and analytes | low | high | |||||||
| Stationary phase, when exposed to organic solvents | instability | ||||||||||
| Loading of analyte into the sorbent | limited | higher | limited to polar compounds | ||||||||
| Thermal stability of physically holding sorbent | low | ||||||||||
| Diffusion of the analytes into viscous sorbents | slow | slow | |||||||||
| Lifetime of the physically holding sorbent | short | ||||||||||
| Matrix Effects | Selective for target analytes | strong | high | decrease analyte recovery | strong | ||||||
| Recovery (of target analyte) | high | sometimes low | high | sometimes low | high | ||||||
| Nº of available stationary phases/Fragility of fiber/Analyte carryover/Sorbent dispersion | limited/yes/possible | impaired by addition of salt; lower at high pH values | |||||||||
| Additional clean-up steps | needed (to reduce the number of interferers | needed (to reduce the number of interferers | |||||||||
| Capacity and dispersibility of sorbent in liquid samples | High |
| Antibiotics Residues Analyzed | Detector | Conditions | Analytical Column | Internal Standard | Ccalfa and Ccbeta | References |
|---|---|---|---|---|---|---|
| PP | UHPLC-MS/MS (Acquity UPLC) coupled to a TSQ Quantum Access Max triple-quadrupole MS, (Thermo Fisher, San Jose, CA, USA) | Mobile phase: A- 50 mL ACN + 3 mL of FA + 0.1 mL of TCA into a 1000 mL volumetric flask and diluted to volume with purified water. B- 50 mL of purified water + 3 mL of FA + 0.1 mL of TCA into a 1000-mL volumetric flask and diluted to volume with ACN. Gradient program: 0–2 min with 8% B and flow 0.4 mL/min, 2–7 min with 8–20% B, 7–8 min with 20–30% B, 8–11 min with 30–100% B, 11–11.1 min with 100% B and flow 0.4–0.8 mL/min, 11.1–12.5 min with 100%, 12.5–12.51 min with 100–8% B and flow 0.8–0.4 mL/min, 12.51–14 min with 8% B. Flow-rate: 0.4 mL/min Ionization: ESI source in the positive mode Temperature column: 30 °C Injection volume: 20 μL Capillary voltage: 3 kV Collision gas (argon) pressure: 1.5 mtorr Vaporizer temperature: 380 °C Sheath gas pressure: 45 units Auxiliary gas flow: 10 units | Kinetex C-18 column (2.1 × 150 mm × 2.6 µm) with an installed pre-filter (Krudkatcher), both from Phenomenex (Torrance CA, USA) | Muscle LOD (µg/kg) = 5–30 CCα (µg/kg) = 8.6–177.5 CCβ (µg/kg) = 14.7–251.33 Kidney LOD (µg/kg) = 15–30 CCα (µg/kg) = 9.5–241.8 CCβ (µg/kg) = 16.2–358.5 | [87] | |
| BZM, AT | UHPLC-MS/MS (Acquity UPLC) coupled to a TSQ Quantum Ultra AM triple-quadrupole MS (Thermo Fisher, San Jose, CA, USA) equipped with electrospray (ESI), atmospheric pressure chemical ionization (APCI) and atmospheric pressure photoionization (APPI) | Mobile phase: A- 0.1% FA; B-ACN Gradient program: first, an isocratic step at 5% solvent B was held for 0.7 min and then the organic modifier percentage increased to 25% during 1.3 min, in a third stage solvent B was raised to 40% in 2 min and to 100% in 4 min more and held for 1 min at this percentage before returning to the initial settings Column temperature: 25 °C Flow rate: 500 µL/min Injection volume: 10 μL Ion-transfer tube temperature: 200 °C APCI Current discharge: 15 µA Vaporizer temperature: 300 °C Voltage: 4 kV for ESI; 10 eV (krypton lamp) for APPI Dwell time: 50 ms Collision gas: 1.5 mTorr Collision energy (CE): 5–35 eV Data acquisition: selected reaction monitoring (SRM) | Ascentis Express C18 column (150 mm × 2.1 mm, 2.7 µm, superficially porous particles) from Supelco | Mebendazole-d3 | LOD (µg/Kg) = 1–10 LOQ (µg/Kg) = 0.6–1.5 | [70] |
| AM, AV, BZM, BA, PN, CP, DYE, IO, LS, MA, NMZ, NSAIDs, PP, AM, QN, SA, TC, Miscellaneous, Flukacides (150) | LC-MS/MS coupled to a triple quadrupole time-of-flight (Q-TOF) 6530 MS (Agilent, Santa Clara, CA, USA) | Mobile phase: 0.1% FA (A) and ACN (B) Gradient program: 5% ACN for 2 min, ramp 5−50% ACN over 10 min, hold at 50% ACN for 1 min, ramp 50−100% ACN over 3 min, hold at 100% ACN for 2 min. For most analytes, the column was held at 100% ACN for the 2 min described above for a total analysis time of 18 min. However, some compounds being investigated did not elute in that time frame, so an additional 4 min of 100% ACN can be added to the chromatographic program to make an extended LC program that is 23 min long Flow rate: 0.25 mL/min Ionization: ESI Agilent Jet Stream Technology Column temperature: 22 °C Injection volume: 10 μL Fragmentor: 150 V, nozzle: 250 V, Vcap: 4000 V, drying gas (N2): 11 L/min, sheath gas (N2): 11 L/min TOF (MS1 only) data acquisition rate: 4 GHz (to m/z 1700); Scan range: m/z 100−1200 at 1.08 spectra/s Compounds that ionized in the negative ion mode were analyzed separately using a different acquisition program. The MS source parameters were as above for negative ions except that the Vcap and nozzle voltages were 2000 and 200 V, respectively. MS/MS data were collected at three different collision energies (CEs) for each compound. The formula “[3 × (mass/100)] + 10” is suggested by Agilent to calculate a reasonable CE for compounds using their acquisition software and was used here; data were also collected at approximately 10 V higher and lower than that value. Medium isolation width (4 m/z) and 200 ms/spectrum were used to collect product ion spectra. | YMC ODS-AQ (120 Å, 2 100 mm, 3 μm) from Waters Corp. (Milford, MA, USA). | Chloramphenicol-d5 | Estimated screening, LOD—10–100 ng/L | [107] |
| MA, LS | LC–MS/MS, LC (Agilent 1100 Series) and an API 5000 MS (AB Sciex, Foster City, CA). | Mobile phase: A- 0.1% FA; B- ACN with 0.1% FA Gradiente program: starts keeping 98% of A during 1 min, and then decreasing linearly to 5% of A during 4 min. This condition is held for 3 min. Finally, A% increases linearly until 10 min, returning to 98%, and this condition is kept for 2 min, with a total run time of 12 min. Between each analysis, 3 min of equilibration time is applied, using the initial gradient conditions (98% A) Flow rate: 0.3 mL/min Source temperature: 550 °C Curtain gas (CUR): 20 ps Ion spray voltage (IS): 5500 V Ion source gas 1 (GS1): 55 psi, Gas 2 (GS2): 45 psi Interface Heater on Collision gas (CAD): 4 mTorr Entrance potential (EP): 10 V Dwell time: 100 ms Data acquisition: MRM mode | HPLC column AgellaDurashell RP (100 mm × 2.1 mm, 5 μm); guard column filled with C18 (4.0 mm × 3.0 mm, 5 μm, from Phenomenex) | Milk LOD (µg/L) = 5–25 LOQ (µg/L) = 10.0–50.0 CCα (µg/L) = 50.2–230.0 CCβ (µg/L) = 60.5–272.1 Muscle (Bovine) LOD (µg/kg) = 6.2–12.5 LOQ (µg/kg) = 12.5–25.0 CCα (µg/kg) = 57.9–120.9 CCβ (µg/kg) = 65.7–149.5 | [31] | |
| Amoxicillin and metabolites | LC-MS/MS, Agilent 1200 LC system (Agilent Technologies Inc., Palo Alto, CA, USA) coupled with a 6460 triple quadrupole MS (Agilent, Palo Alto, California, USA) | Mobile phase: A- ACN; B- 0.1% FA in water Gradient program: time 0, 100% B; 2 min, 98% B; 5 min, 80% B; 12 min, 50% B; 20 min, 98% B Flow rate: 1 mL/min Injection volume: 20 μL Ionization: ESI source in positive mode Capillary voltage: 4 kV Gas temperature: 300 °C Gas flow: 10 L/min Nebulizer gas: 15 ps Sheath gas temperature: 250 °C Sheath gas flow: 7 L/min Data acquisition: MRM mode | Agilent C18 column (50 mm × 4.6 mm; 5 µm) protected with a guard column (C18; 5 µm) | Penicillin V | LOD (µg/kg) = 0.1–0.6 LOQ (µg/kg) = 0.3–0.8 µg/kg CCα (µg/kg) = 11.1–11.15 µg/kg CCβ = 12.1–13.0 µg/kg | [58] |
| PN, SA, TC, MA, LS, QN, AM, PT, DP, Rifamycines (62) | High resolution LC-MS. Thermo Ultimate 3000 High Performance LC system (San Jose, CA, USA). Mass spectrometer Q-Orbitrap (Thermo Scientific, San Jose, CA, USA) was coupled with heated ESI (HESIII) source. | Mobile phase: A- aqueous solution containing 0.1% (v/v) FA; B- MeOH Gradient program: initiated with 5% eluent B for 1 min, continued with linear increase to 95% B in 19 min. This condition was maintained for 5 min. The system returned to 5% B in 1 min and was re-equilibrated for 4 min (run time: 30 min) Flow rate: 0.25 mL/min Column temperature: 30 °C HESI-II temperature: 320 °C Capillary temperature: 300 °C Electrospray voltage: 3.00 kV (positive mode) Sheath and auxiliary gas: 35 and 15 arbitrary units. Acquisition: full scan/dd-MS2; Mass range in full scan was within m/z 150–1200. The data were acquired at a resolution of 70,000 FWHM (m/z 200). | Poroshell 120 EC-C18 column (100 × 3.0 mm; 2.7 µm); guard column Poroshell (2.1 × 5 mm) | Penicillin G-d7 spiramycin I-d3 tetracycline-d6 florfenicol-d3 sulfamethazine-13C6 sulfanilamide-13C6 enrofloxacin-d5 cefadroxil-d4 | Muscle (bovine) CCα (µg/kg) = 1–1128.0 CCβ (µg/kg) = 3.3 -1272 Muscle (swine) CCα (µg/kg) = 1–1374 CCβ (µg/kg) = 3.3–1573 Muscle (poultry) CCα (µg/kg) = 1–441 CCβ (µg/kg) = 3.3–486 | [108] |
| AG, PN, MA, SA, AT, CD, NSAIDs, Pharmaceuticals, Rifaximin, Baquiloprim, Trimethoprim, Chlorpromazine, 6-phenyl-2-thiouracil, Bromhexine (76) | UHPLC-MS/MS. Thermo UHPLC Accela system (Thermo, San Jose, CA, USA) coupled to a Thermo Scientific TSQ Quantum Access Triple Quadrupole MS, USA) | Mobile phase: A- ACN, B- aqueous ammonium formate 1 mM with 0.1 FA (v/v), C- MeOH Gradient program: 80% A and 20% B (0%C), decreased linearly to 0% A and 95% B (5%C) in 10 min and was held for additional 4 min. Total run time: 20 min Injection volume: 10 µL Spray voltage: 4000 V Sheath gas: 25 psi Auxiliary gas: 10 a.u. Capillary temperature: 300 °C. Data acquisition: reaction monitoring (SRM) mode | ACQUITY UPLC BEH HILIC (100 mm × 2.1 mm, 1.7 µm, Waters) | Nigericin amikacin flubendazole flunixin d3 meloxicam d3 sulfadiazine d4 sulfadimidine d4 sulfadimethoxine d4 | LOQ (µg/kg) = 0.03–178 CCα (µg/kg) = 2.2–1151 CCβ (µg/kg) = 2.4–1302 | [72] |
| QN, MA, PN, NMZ, SA, LS, AM, QNX, TC, PP, tiamuline, bacitracin, flavomycin, antibacterial synergists, including Diaveridine (DVD), Trimethoprim (TMP). (12 classes of drugs, 120 compounds) | HPLC-MS/MS. Finnigan HPLC system (Thermo Fisher Scientific, Waltham, MA, USA) coupled to TSQ Quantum Access triple quadrupole MS (Thermo Fisher Scientific). | Mobile phase: A- 0.1% FA in water, B- ACN Gradient program: 0–5 min, 0.1 mL/min, 5% B; 5–10 min, 0.2 mL/min, linear increase to 25% B, and hold for 15 min; 25–40 min, 0.2 mL/min, increase to 50% B; 40–45 min, increase to 80% B, hold for 5 min; 50–50.5 min, decrease to 5% B, hold for 9.5 min. Auxiliary gas: N2 at a pressure of 5 arbitrary units Collision gas: Argon used for collision induced dissociation at a pressure of 25 arbitrary units. Column temperature: 40 °C Injection volume: 10 µL Spray voltage: 4.5 KV Capillary temperature: 350 °C | Hypersil Gold C18 (150 × 2.1 mm, 5 µm) column | LOD (µg/kg) = 0.5–3.0 LOQ (µg/kg) = 1.5–10.0 | [96]) | |
| SA, QN | UHPLC-MS/MS. Waters ACQUITY UPLC system was coupled to a Xevo triple quadrupole MS (Waters, Milford, MA, USA). | Mobile phase: A-MEOH; B- 0.1% FA in water Gradient program: 0.0% A; 0–2 min, linear increase 25% A; 2–7 min, linear increase 30% A; 7.01–8 min, 45% A linear increase 60% A, and hold for 3 min; 11.01–12 min, 100% A hold for 1 min; 12–14 min, decrease 10% A, and hold for 2 min. Flow rate: 0.2 mL/min Injection volume: 10 μL Ionization: ESI in the positive mode Capillary voltage: 3 kV Source temperature and desolvation temperature: 150 and 400 °C, respectively. Data acquisition: MRM mode | Waters Acquity UPLC CORTECS C18 column (150 mm × 2.1 mm, 1.6 μm) | LOD (µg/kg) = 0.05–2.6 LOQ (µg/kg) = 0.12–5.6 | [68] | |
| TC (5) and epimers TC’s (5) | LC-MS/MS. Agilent 1290 Infinity LC system (Waldbronn, Germany) coupled to a QTrap 5500 triple quadrupole fitted with the Turbo V™ ion source (Sciex, Toronto, Canada). | Mobile phase: A- 0.01 M oxalic acid, B- MEOH containing 0.1% FA Gradient program: 11-min gradient was set as follows: 0–0.5 min (1% B); 0.5–2 min (35% B); 2–3.5 min (55% B); 3.5–6 min (55% B); 6–6.5 min (100% B); 6.5–8 min (100% B); 8–8.5 min (5% B); 8.5–11 min (5% B). Flow rate: 0.4 mL/min Injection volume: 10 μL Ionization: ESI source in positive mode Temperature (TEM): 550 °C Curtain gas (CUR): 30 psi Nebulising gas (GS1): 40 psi Drying gas (GS2): 40 psi Collision activated dissociation (CAD) gas pressure: Medium. Ion spray voltage: 5000 V Data acquisition: MRM mode | Acquity UPLC HSS T3 column (2.1 × 100 mm, 1.8 µm), guard column: HSS T3 VanGuard pre-column (2.1 × 5 mm, 1.8 µm) | STC (µg/kg) = 50 | [45] | |
| PN, CP, SA, TC, MA, LS, QN, AM, PT, PP, DP, Novobiocin, Rifaximin, Virginiamycin M1 (75) | LC-MS/MS Shimadzu LC20AD-XR system (Kyoto, Japan) coupled to API4000 MS (ABSciex, San Jose, CA, USA) | Mobile phase: A-mixture of 1 mmol/L HFBA and 9.5 mmol/L PFPA in water; B- mixture of 1 mmol/L HFBA and 9.5 mmol/L PFPA in ACN Gradient program: started with 10% B. It was then raised to 30% B over 4 min, then held for 1 min and again raised linearly to 70% B over 2 min and held for 3 min. The initial composition was then recovered over a 1-min delay. Flow rate: 0.60 mL/min Ionization: ESI source in positive mode Source temperature: 700 °C Turbo-ion-spray voltage: 5500 V Sheath gas pressure (air): 40 psi Auxiliary gas pressure (air): 50 psi Curtain gas: 20 psi Data acquisition: MRM mode | Waters Symmetry C18 column (150 mm × 3.9 mm, 5 µm), protected with a C18 security guard system from Phenomenex (4.0mm × 3.0mm, 5 µm) | Sulfaphenazole | Cval (µg/kg) = 50–400 | [52] |
| TT, NMZ, LS, AT, TC, PN, QN, BA, CP, SA, MA, AM, CDC, TQ, Flukicide, NSAID, Progestin | UHPLC-MS/MS. UHPLC system was coupled to a triple quadrupole MS model 6490 system (Agilent Technologies, Santa Clara, CA, USA) equipped with electrospray Jet Stream, and iFunnel technology. | Mobile phase: A- 0.1% FA in water; B- 0.1% FA in ACN Gradient program: 10% B for 0–0.5 min, then to 100% B by 8 min, and hold 100% B until 12 min, followed with a post column equilibrium time for 3 min Flow rate: 0.3 mL/min Column temperature: 30 °C Injection volume: 3 µL Gas temp: 120° C Gas flow: 14 L/min Capillary voltage: 3000 V Nebulizer pressure: 40 psi Sheath gas heater: 400 °C Sheath gas flow: 12 L/min Nozzle voltage: 0 V for both positive and negative ion mode iFunnel parameters were: high pressure RF of 90 V for positive and negative ion mode; and low pressure RF of 70 V for positive and 60 V for negative ion mode Data acquisition: MRM | Poroshell 120 EC-C18 column (150 × 2.1 mm, 2.7 µm), Poroshell 120 guard column (5 × 2.1 mm, 2.7 µm) (Agilent, Newport, DE, USA) | Flunixin-d3 (NEG) Flunixin-d3 (POS) | LOQ (µg/kg) = 1–5 | [51] |
| AM, AV, BZM, CCD, LS, MA, NSAIDs, QN, SA, TQ, Other antibiotics (105) | LC-MS/MS. Agilent 1290 Infinity LC system (Waldbronn, Germany) coupled to a QTrap 5500 triple quadrupole (Sciex, Toronto, Canada) | Mobile phase: A- water, B- MEOH) both containing 0.5 mM of ammonium formate and 0.1% FA Gradient program: 1- Multifamily run (16 min): 0–0.5 min: 5% B; 0.5–2.5 min: linear gradient to 35% B; 2.5–10 min: linear gradient to 100% B; hold at 100% B for 2 min; return to 5% B in 0.5 min and hold at 5% B for 3.5 min Flow rate: 0.4 mL/min Injection volume: 10 µL LC flow directed into the MS detector between 0.5 and 12 min using the integrated diverter valve Ion source: 550 °C Curtain gas (CUR): 30 psi Nebulising gas (GS1): 40 psi Drying gas (GS2): 40 psi Collision activated dissociation (CAD) gas pressure set as medium Ion spray voltages (IS): 5000/−4500 V. Gradient program: 2- Avermectins run (10 min): 0–0.2 min: 5% B; 0.2–1 min: linear gradient to 75% B; 1–4.5 min: linear gradient to 100% B; hold at 100% B for 2 min; return to 5% B in 0.5 min and hold at 5% B for 3 min. Elution conditions: 0.4 mL/min Injection volume: 10 µL Avermectins Ion source: 350 °C The other parameters were kept. Data acquisition: MRM mode | Acquity BEH VanGuard pre-column (2.1 × 5 mm, 1.7 µm) and Acquity BEH C18 LC column (2.1 × 100 mm, 1.7 µm) (both from Waters, BadenDättwil, Switzerland) | STC (µg/Kg) = 0.3–10 | [98] | |
| AV, BZM, BA, CP, CAP, FF, DYE, HOM, IZ, MA, QN, QX, TQ, SA, TC, organochlorines, organophosphates, others (>200) | High resolution LC-MS. Ultrahigh-performance liquid chromatography (UHPLC) system (DionexUltiMate 3000, Thermo Fisher Scientific) coupled to a quadrupole-Orbitrap MS with ESI (Q-Exactive, Thermo Fisher Scientific) | Mobile Phase:A- 0.1% FA in ACN, B- 0.1% FA in water Gradient program: 5% (A) 0.1% FA in ACN for 3 min, then linearly increased to 100% in 19min, and kept constant for 3 min. In the end, the eluent was restored to the initial conditions for 5 min to re-equilibrate Flow rate: 0.3 mL/min Injection volume: 10 µL Ionization: ESI in positive and negative mode Spray voltage: 3200 V (positive mode), 2800 V (negative mode) Sheath gas flow rate: 8 L/min Auxiliary gas flow rate: 10 L/min Sweep gas flow rate: 1.5 L/min Capillary temperature: 325 °C S-lens RF level: 60 V Data acquisition: Full MS/ddMS2 (with inclusion list/full-scan data dependent MS/MS) mode over the mass range m/z 100–1000 (positive mode) and 150–1000 (negative mode) | Accucore RP-MS C18 column (2.1 × 100 mm, 2.6 μm, Thermo Fisher Scientific, USA) | SL (carp, shrimp, crab, eel, mussel) (µg/kg) = 1–50 | [106] | |
| AM (CAP, FF, TAP) | LC-MS/MS. Agilent 1200 Series HPLC (Agilent Technologies Inc., Santa Clara, CA, USA) coupled to a Triple Quadrupole MS detector QTRAP API 5500 Sciex (Framingham, MA, USA) | Mobile phase: A- 0.1% of FA in water, B- 0.1% of FA acid in MEOH Gradient program: 90% A; 0.5 min–90% A; 2.0 min–70% A; 7.0 min–90% A; and 9.0 min–90% A; Total run time: 9 min. Flow rate: 400 µL/min Injection volume: 10 µL Column temperature: 40 °C | Aqua C18 column (50 × 2.0 mm, 5.0 μm, Phenomenex, Inc., Torrance, USA) | Deuterated chloramphenicol (CAP-d5) | LOQ (µg/kg) = 0.15–12.5 CCβ (μg/kg) = 0.068–58.91 | [76] |
| AG, PP | High- resolution LC-MS. Thermo Ultimate 3000 High Performance LC system (Thermo Scientific, San Jose, CA, USA) coupled with mass spectrometer Q-Orbitrap (Q-Exactive, Thermo Scientific, San Jose, CA, USA) equipped with heated ESI (HESI-II) source. | Mobile phase: A- aqueous solution containing 1% (v/v) FA (FA) and 1mM ammonium formate (AF), B- ACN Gradient program: 20% eluent A for 2 min, continued with linear increase to 35% A in 5 min. In 1 min eluent A increased to 95%, and this condition was maintained for 7 min. The system returned to 20% B in 0.1 min and was re-equilibrated for 4 min (run time: 17 min). Column temperature: 40 °C Flow rate: 0.25 mL/min Injection volume: 5 µL HESI-II temperature: 300 °C Capillary temperature: 250 °C Electrospray voltage: 3.00 kV (positive mode); S-lens value: 50 V Sheath and auxiliary gas: 35 and 25 arbitrary units, respectively. | InfinityLab Poroshell 120 HILIC column (100 × 2.1 mm, 2.7 µm, Agilent Technologies, Santa Clara, CA, USA) connected with the InfinityLab Poroshell 120 HILIC guard column (5 × 2.1 mm, 2.7 µm) | Ribostamycin | Muscle CCα (µg/kg) = 10–1164 CCβ (µg/kg) = 10–1355 Milk CCα (µg/kg) = 10–167 CCβ (µg/kg) = 10–1860 | [88] |
| SA, PN, CP, TT, PT, AM, LC, MA, DP, QN (60) | LC-MS/MS. Surveyor LC pump, coupled with a triple quadrupole mass spectrometer (TSQ Quantum Ultra, Thermo Fisher, San Jose, CA, USA) | Mobile phase: A- aqueous solution 0.1% (v/v) FA; B- MEOH Flow-rate: 0.25 mL/min Gradient program: 2% B for 1 min and increased linearly up to 95% B in 19.5 min; this condition was maintained for 5 min before returning to initial condition in 1 min (2% B) and held for 4 min to equilibrate the column. Ionization: ESI source in positive mode Column temperature: 30° C Injection volume: 10 µL Capillary temperature: 300 °C Vaporizer: 320 °C Spray voltage: 3 kV Sheath gas pressure: 35 units Auxiliary gas (nitrogen) pressure: 35 units Collision gas (argon) pressure: 1.5 mtorr Data acquisition: selected reactions transitions (SRM) monitored | Poroshell 120 EC-C18 column (3.0 × 100 mm; 2.7 µm). Guard column Poroshell (2.1 × 5 mm), both from Agilent Technologies (Santa Clara, CA, USA) | sulfanilamide-13C6 sulfamethazine-13C6 enrofloxacin-d5 florfenicol-d3 spiramycin-d3 metacycline | Muscle (bovine, porcine, ovi-caprine, rabbit, equidae) CCα (µg/kg) = 10 -1159 CCβ (µg/kg) = 10 -1349 Muscle (poultry) CCα (µg/kg) = 10 -448 CCβ (µg/kg) = 10 -502 Muscle (aquaculture) CCα (µg/kg) = 10 -1262 CCβ (µg/kg) = 10 -1594 Milk CCα (µg/kg) = 10 -239 CCβ (µg/kg) = 10 -286 | [86] |
| AM, AT, AV, BA, CP, CCD, IO, LS, NMZ, NSAIDs, PN, QN, SA, TC, TQ, QNX, CDC, Insecticides, Estrogens, Androgens, Other antibiotics (164) | High resolution UHPLC-MS. Dionex UltiMate 3000 UHPLC system (Thermo Fisher Scientific, San Jose, CA, USA) coupled with Q-Orbitrap HRMS (Thermo Fisher Scientific) | Mobile phase: (A) 0.1% FA in water, (B) 0.1% FA in ACN, and (C) 0.1% FA in MEOH Gradient program: Flow rate: 0.2 mL/min: 90% of solvent A and 10% C (0% B) 0 min., 90% of solvent A and 10% C (0% B) (0.5 min.); 30% of solvent A and 70% C (0% B) (4 min); Flow rate: 0.3 mL/min: 20% of solvent A, 10% B and 70% C (5 min); 20% of solvent A, 70% B and 10% C (12 min); 0% of solvent A, 70% B and 30% C (15 min); 0% of solvent A, 70% B and 30% C (18 min.); Flow rate: 0.2 mL/min: 90% of solvent A, 0% B and 10% C (20 min); 90% of solvent A, 0% B and 10% C (25 min). Injection volume: 10 µL Column temperature: 40 °C Ionization: Electrospray voltage 4.0 kV in positive and 3.5 kV in negative ionization modes Heater temperature 350 °C Capillary temperature: 300 °C Sheath gas (N2): 20 arbitrary units (arb) Auxiliary gas (N2): 6 arb, S-lens RF: 50 arb. Data acquisition: Full scan data both in the positive and negative ionisation modes Resolving power: 70,000 FWHM | Phenomenex Luna Omega C18 analytical column (100 × 2.1 mm, 1.6 µm) | Albendazole-d3 Azaperone-d4 Chloramphenicol-d5 Cyromazine-d4 Diclofenac-13C6 DNC-d8 Enrofloxacin-d5 Flunixin-d3 Megestrole acetate-d3 Nigericin Toltrazuril-d3 Triclabendazole-d3 β-Testosterone-d2 | CCβ (µg/kg) = 0.038–547.0 | [101] |
| AM (CAP, FF, TAP) | LC-TOF-MS. Agilent 1290 Infinity II Series HPLC (Agilent Technologies, Santa Clara, CA, USA) coupled to an Agilent 6550 QTOF mass spectrometer, using an Agilent jet stream dual ESI (AJS-Dual ESI) interface. | Mobile phase: 70:30 water:MeOH (v/v) mixture under isocratic conditions Flow rate: 0.4 mL/min Ionization: ESI in negative mode Nebulizer gas pressure: 30 psi Drying gas: 130 °C Sheath gas: 300 °C Capillary spray, nozzle, fragmentor and octopole 1 RF Vpp voltages: 4000, 500, 360 and 750 V, respectively Collision energy: 40 V carried out with 2 GHz extended dynamic range mode and 3 spectra/s, 333.3 ms/spectrum and 1999 transients/spectrum. Reference mass of 112.985587 and 922.0098 | Zorbax RRHD Eclipse Plus C18 (100 × 2.1 mm, 1.8 μm, Agilent) | LOD (pg/L) = 29.6 (TAP) LOD (pg/L) = 3 (FF) LOD (pg/L) = 3 (CAP) | [109] | |
| PN, QN, MA, SA, TC, AM, DP | UHPLC-MS/MS. UHPLC system was coupled with an Agilent 6460 triple quadrupole tandem MS | Mobile phase: A- 0.1% FA solution, B- ACN acidified with 0.1% FA. Gradient mode: starting with 20% B for 2 min, increasing linearly until 90% over 8 min, remaining constant for 2 min and then returning to the initial proportion. A post-run interval of 5 min was necessary to re-equilibrate the column to the initial conditions. Flow rate: 0.5 mL/min Column temperature: 30 °C Injection volume: 20 μL Ionization: ESI source in the negative mode for the amphenicols and in the positive mode for the remaining compounds Source Temperature: 300 °C Gas flow:12 L/min Nebulizer gas: 30 psi Sheath gas flow: 12 L/min Sheath gas temperature: 350 °C Capillary voltage: 4.0 kV (positive mode) and 3.5 kV (negative mode) Nozzle: 1.5 kV Data acquisition: MRM mode | Agilent Zorbax SB C18 (4.6mm × 150 mm, 3.5 μm) column (Agilent Technologies, CA, USA) | 1-penicillin G-d7 2-enrofloxacin-d5 3-roxythromycin 4-sulfadoxine-d3 5-chloramphenicol-d5 6-Oxytetracycline 7-Sulfathiazole 8-Enrofloxacin-d5, at concentration 50 ng/g | LOD (µg/kg) = 0.1–0.3 LOQ (µg/kg) = 0.3–10 CCα (µg/kg) = 0.37–1008.34 CCβ (µg/kg) = 0.44–1016.68 | [37] |
| PN (amoxicillin) | LC-MS/MS. Shimadzu LC system (LC-30CE; Japan) consisted of a triple quadrupole MS (AB SCIEX Triple Quad™ 5500, AB SCIEX Corp., Framingham, MA, USA) | Mobile phase: A- 0.1% FA in water; B- 0.1% FA in ACN Gradient mode: 0–1.5 min, 95% A; 1.5−3 min, 70% A; 3–3.5 min, 10% A; 3.5−6 min, 95% A Gradient elution: 0.3 mL/min Column temperature: 30 °C Injection volume: 5 μL Ionisation: ESI in positive mode Spray voltage: 5500 V Temperature of ion source: 600 °C Pressures of spray gas: 60 psi Auxiliary gas: 60 psi Curtain gas: 35 psi Data acquisition: MRM mode | Luna Omega C18 column (50 mm × 2.1 mm, 1.6 μm) | Amoxicillin-D4 | Muscle, Liver, Fat, Kidney LOD (µg/kg) = 5 LOQ (µg/kg) = 10 | [81] |
| SA, TQ, NMZ, QN, PT, PN, BZM (78) | UHPLC-MS/MS. Waters Acquity ultra performance liquid chromatography system coupled to Micromass Xevo TQ-S triple quadrupole MS (Waters, Manchester, UK) | Mobile phase: A- 0.1% FA, B- MeOH:ACN, 2:8, v/v, containing 0.1% FA Gradient program: 0–1.0 min, 97% A; 1.0–4.5 min, 97–83% A; 4.5–6.0 min, 83–55% A; 6.0–6.5 min, 55–0% A; 6.5–7.5 min, 0% A; 7.5–7.6 min, 0–97% A; 7.6–9.5 min, 97% A Elution conditions: 0.3 mL/min Capillary voltage: 2.5 kV Source temperature: 150 °C Desolvation temperature: 500 °C Desolvation gas flow rate: 800 L/h Cone gas flow rate: 150 L/h Collision gas: argon at 0.15 mL/min Data acquisition: MRM mode | Acquity UPLC BEH C18 column (100 × 2.1 mm, 1.7 μm; Waters, Milford, MA, USA) | LOQ (µg/kg) = 0.1–1 | [105] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barros, S.C.; Silva, A.S.; Torres, D. Multiresidues Multiclass Analytical Methods for Determination of Antibiotics in Animal Origin Food: A Critical Analysis. Antibiotics 2023, 12, 202. https://doi.org/10.3390/antibiotics12020202
Barros SC, Silva AS, Torres D. Multiresidues Multiclass Analytical Methods for Determination of Antibiotics in Animal Origin Food: A Critical Analysis. Antibiotics. 2023; 12(2):202. https://doi.org/10.3390/antibiotics12020202
Chicago/Turabian StyleBarros, Sílvia Cruz, Ana Sanches Silva, and Duarte Torres. 2023. "Multiresidues Multiclass Analytical Methods for Determination of Antibiotics in Animal Origin Food: A Critical Analysis" Antibiotics 12, no. 2: 202. https://doi.org/10.3390/antibiotics12020202