
QSAR Studies, Molecular Docking, Molecular Dynamics, Synthesis, and Biological Evaluation of Novel Quinolinone-Based Thiosemicarbazones against Mycobacterium tuberculosis
 , , , , , , , , , and
, , , , , , , , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. QSAR Modeling
2.2. Applicability Domain (AD)
2.3. External Validation of the Model QSAR
2.4. Design of the Structures
2.5. Molecular Orbitals
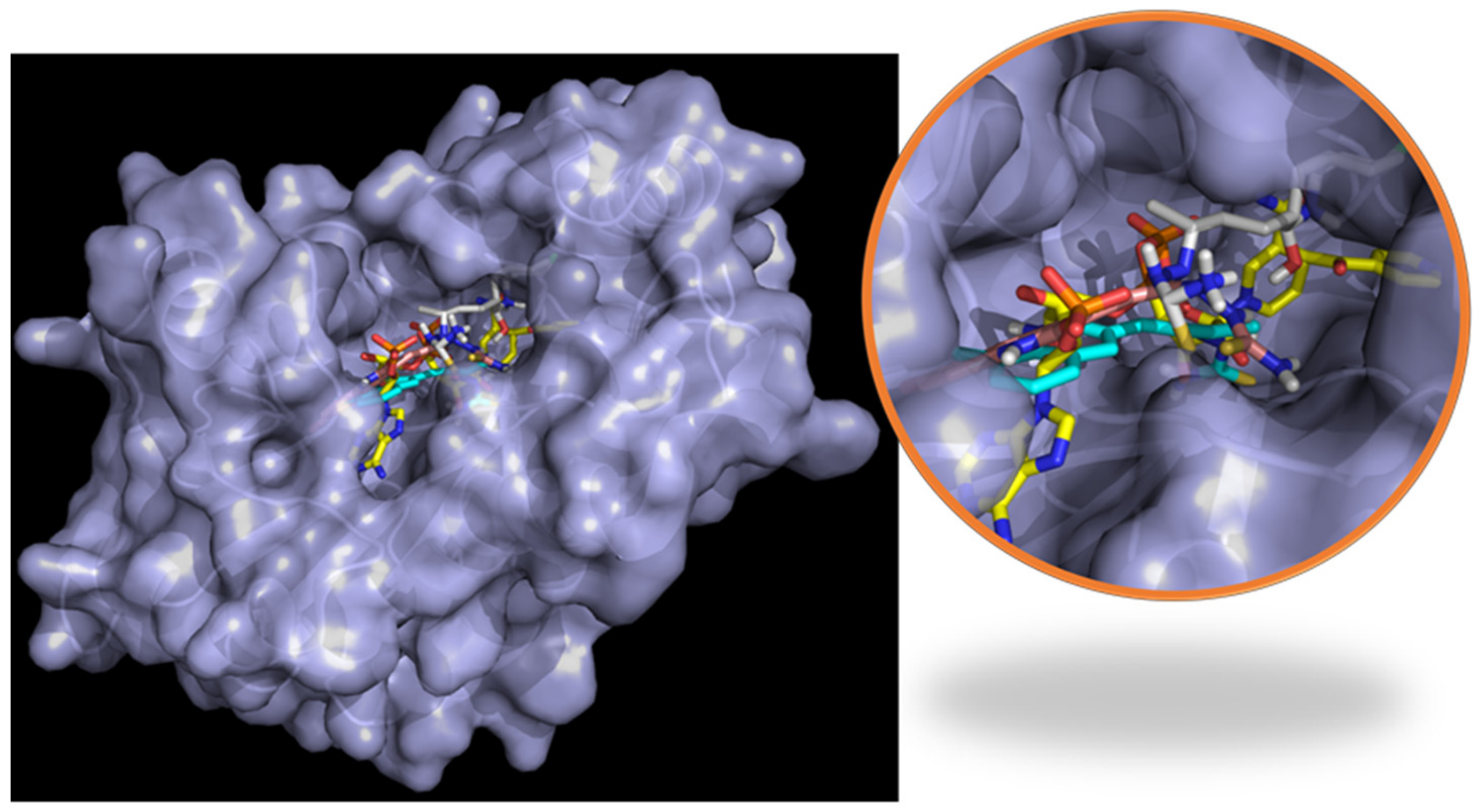
2.6. Molecular Docking
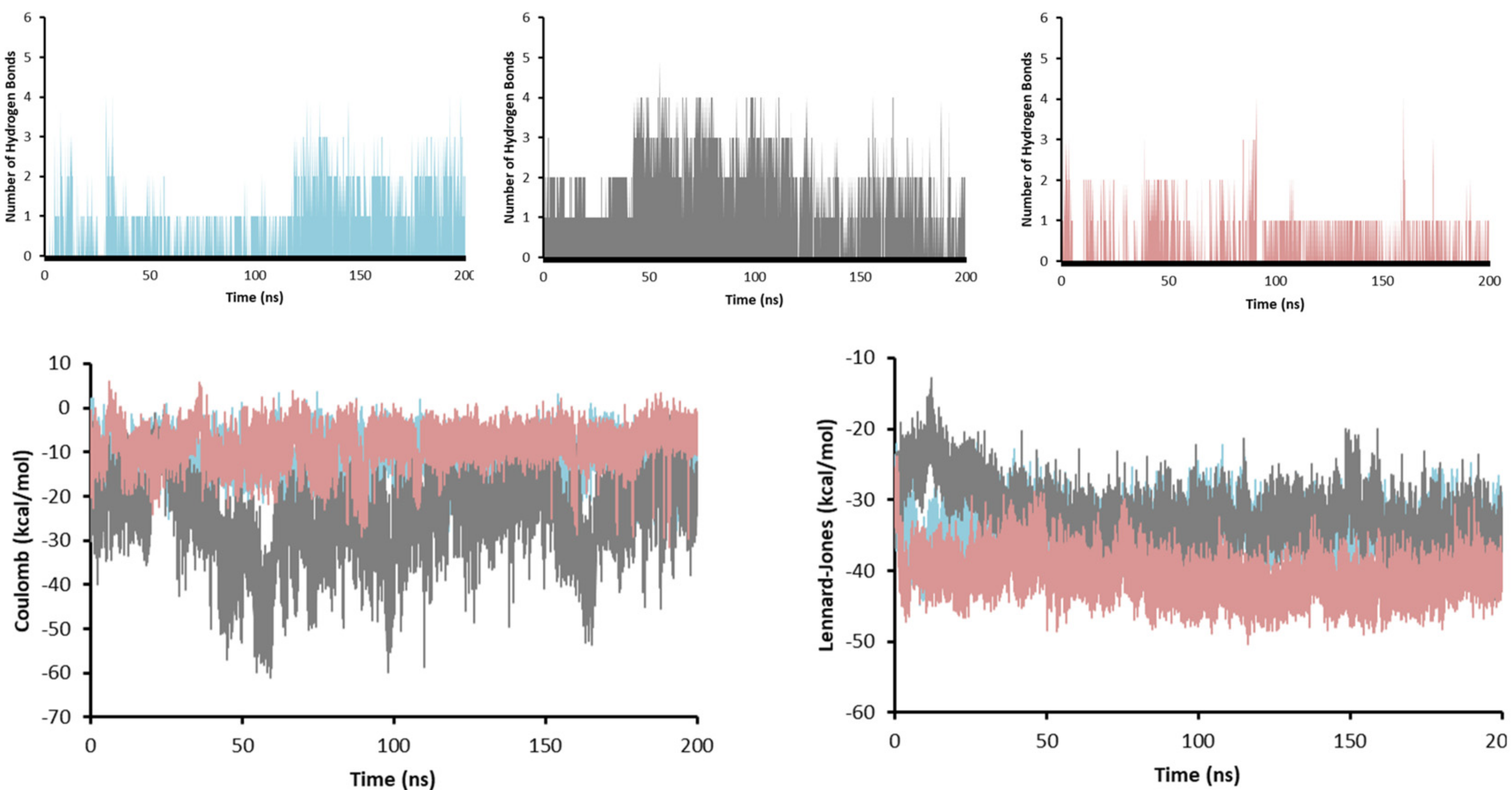
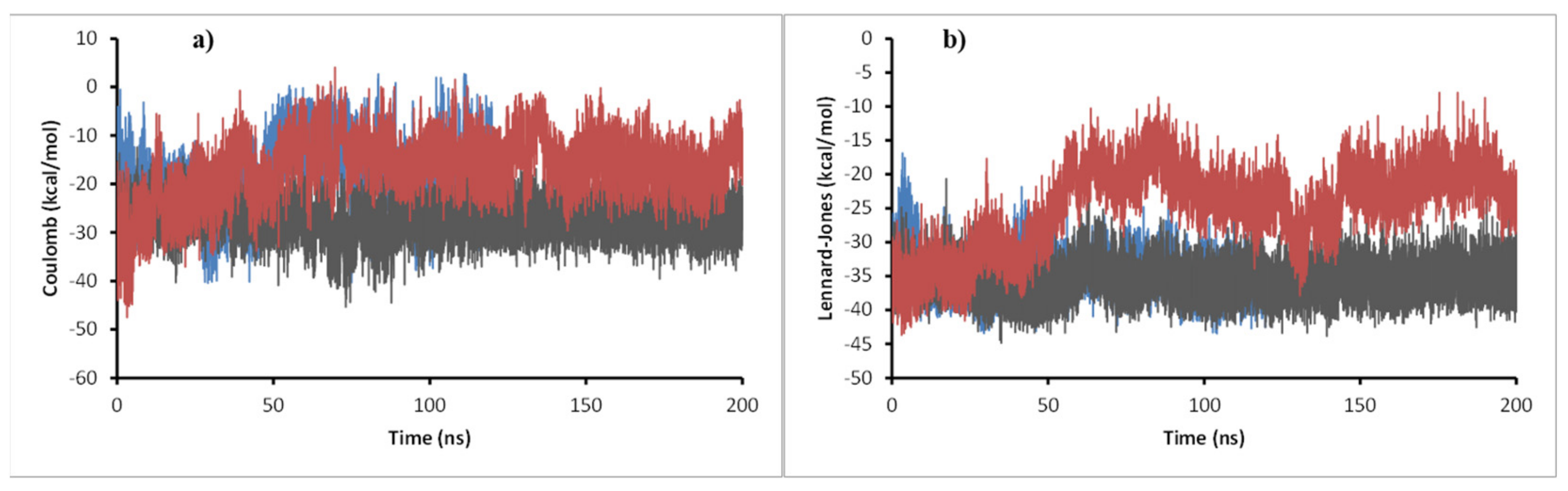
2.7. Molecular Dynamics
2.8. Chemistry
2.9. Antimycobacterial Activity
2.10. Cytotoxicity
3. Materials and Methods
3.1. Data Collection
3.2. Minimum Energy Structures and Molecular Descriptors Calculation
3.3. Descriptor Calculations
3.4. QSAR Models
3.5. Applicability Domain (AD)
3.6. Validation Procedures
3.7. Molecular Docking
3.8. Molecular Dynamics
3.9. Molecular Orbitals
3.10. Chemistry
3.11. Antimycobacterial Activity
3.12. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dagne, B.; Desta, K.; Fekade, R.; Amare, M.; Tadesse, M.; Diriba, G.; Zerihun, B.; Getu, M.; Sinshaw, W.; Seid, G.; et al. The Epidemiology of First and Second-Line Drug-Resistance Mycobacterium Tuberculosis Complex Common Species: Evidence from Selected TB Treatment Initiating Centers in Ethiopia. PLoS ONE 2021, 16, e0245687. [Google Scholar] [CrossRef] [PubMed]
- Visca, D.; Ong, C.W.M.; Tiberi, S.; Centis, R.; D’Ambrosio, L.; Chen, B.; Mueller, J.; Mueller, P.; Duarte, R.; Dalcolmo, M.; et al. Tuberculosis and COVID-19 Interaction: A Review of Biological, Clinical and Public Health Effects. Pulmonology 2021, 27, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Kranzer, K.; Khan, P.; Godfrey-Fausset, P.; Ayles, H.; Lönnroth, K. Tuberculosis Control. Lancet 2016, 387, 1159–1160. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Dorman, S.E. Latent Tuberculosis Infection. N. Engl. J. Med. 2021, 385, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Beena, P.M.; Devnikar, A.V.; Mali, S. A Systemic Review on Tuberculosis. Indian J. Tuberc. 2020, 67, 295–311. [Google Scholar] [CrossRef] [PubMed]
- The Global Fund to Fight AIDS Global Fund Applauds Japan’s Major Commitment to Help End AIDS, Tuberculosis and Malaria and Strengthen Systems for Health. Available online: https://www.theglobalfund.org/en/news/2022/2022-08-27-global-fund-applauds-japans-major-commitment-to-help-end-aids-tuberculosis-and-malaria-and-strengthen-systems-for-health/ (accessed on 1 September 2022).
- Lobo, N.; Brooks, N.A.; Zlotta, A.R.; Cirillo, J.D.; Boorjian, S.; Black, P.C.; Meeks, J.J.; Bivalacqua, T.J.; Gontero, P.; Steinberg, G.D.; et al. 100 Years of Bacillus Calmette–Guérin Immunotherapy: From Cattle to COVID-19. Nat. Rev. Urol. 2021, 18, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Iradukunda, A.; Ndayishimiye, G.-P.; Sinarinzi, D.; Odjidja, E.N.; Ntakaburimvo, N.; Nshimirimana, I.; Izere, C. Key Factors Influencing Multidrug-Resistant Tuberculosis in Patients under Anti-Tuberculosis Treatment in Two Centres in Burundi: A Mixed Effect Modelling Study. BMC Public Health 2021, 21, 2142. [Google Scholar] [CrossRef]
- Ali, A.; Ali, A.; Salahuddin; Bakht, M.A.; Ahsan, M.J. Synthesis and Biological Evaluations of N -(4-Substituted Phenyl)-7-Hydroxy-4-Methyl-2-Oxoquinoline-1(2 H )-Carbothioamides. Polycycl. Aromat. Compd 2021, 42, 4910–4921. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2021; WHO: Geneva, Switzerland, 2021; ISBN 978-92-4-003702-1.
- Allué-Guardia, A.; García, J.I.; Torrelles, J.B. Evolution of Drug-Resistant Mycobacterium Tuberculosis Strains and Their Adaptation to the Human Lung Environment. Front. Microbiol. 2021, 12, 612675. [Google Scholar] [CrossRef]
- Mase, S.R.; Chorba, T. Treatment of Drug-Resistant Tuberculosis. Clin. Chest Med. 2019, 40, 775–795. [Google Scholar] [CrossRef]
- Adeniji, S.E.; Uba, S.; Uzairu, A. Theoretical Modeling for Predicting the Activities of Some Active Compounds as Potent Inhibitors against Mycobacterium Tuberculosis Using GFA-MLR Approach. J. King Saud Univ.Sci. 2018, 31, 1151–1166. [Google Scholar] [CrossRef]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Front. Pharmacol. 2018, 9, 1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braga, R.; Alves, V.; Silva, A.; Nascimento, M.; Silva, F.; Liao, L.; Andrade, C. Virtual Screening Strategies in Medicinal Chemistry: The State of the Art and Current Challenges. Curr. Top. Med. Chem. 2014, 14, 1899–1912. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Santos, S.; Ventura, C.; Elvas-Leitão, R.; Santos, L.; Vitorino, S.; Reis, M.; Miranda, V.; Correia, H.F.; Aires-de-Sousa, J.; et al. Design, Synthesis and Biological Evaluation of Novel Isoniazid Derivatives with Potent Antitubercular Activity. Eur. J. Med. Chem. 2014, 81, 119–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo-Filho, C.C.; Braga, R.C.; Andrade, C.H. 3D-QSAR Approaches in Drug Design: Perspectives to Generate Reliable CoMFA Models. Curr. Comput. Aided Drug Des. 2014, 10, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Hadni, H.; Mazigh, M.; El Hallaoui, M. QSAR and Molecular Docking Studies of 4-Anilinoquinoline-Triazine Hybrids as Pf-DHFR Inhibitors. Mediterr. J. Chem. 2019, 8, 84–93. [Google Scholar] [CrossRef]
- Patel, S.R.; Gangwal, R.; Sangamwar, A.T.; Jain, R. Synthesis, Biological Evaluation and 3D-QSAR Study of Hydrazide, Semicarbazide and Thiosemicarbazide Derivatives of 4-(Adamantan-1-Yl)Quinoline as Anti-Tuberculosis Agents. Eur. J. Med. Chem. 2014, 85, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Nagabushan, H.; Roopadevi, H.S. Bedaquiline: A Novel Antitubercular Drug for Multidrug-Resistant Tuberculosis. J. Postgrad. Med. 2014, 60, 300. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Ahn, S.; Hwang, N.Y.; Jeon, K.; Kwon, O.J.; Huh, H.J.; Lee, N.Y.; Kim, C.-K.; Koh, W.-J. Limited Effect of Later-Generation Fluoroquinolones in the Treatment of Ofloxacin-Resistant and Moxifloxacin-Susceptible Multidrug-Resistant Tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e01784-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Q.; Tao, L.; Li, Y.; Chen, N.; Chen, H.; Zhou, Y.; Wang, Y.; Chen, H.; Tang, Q.; Wang, X.; et al. High-Dose Gatifloxacin-Based Shorter Treatment Regimens for MDR/RR-TB. Int. J. Infect. Dis. 2022, 115, 142–148. [Google Scholar] [CrossRef]
- Alós, J.-I. Quinolonas. Enferm. Infecc. Microbiol. Clínica 2009, 27, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.D.; More, U.A.; Parkale, D.; Aminabhavi, T.M.; Gadad, A.K.; Nadagouda, M.N.; Jawarkar, R. Design, Synthesis of Quinolinyl Schiff Bases and Azetidinones as Enoyl ACP-Reductase Inhibitors. Med. Chem. Res. 2015, 24, 3892–3911. [Google Scholar] [CrossRef]
- Jain, P.P.; Degani, M.S.; Raju, A.; Anantram, A.; Seervi, M.; Sathaye, S.; Ray, M.; Rajan, M.G.R. Identification of a Novel Class of Quinoline–Oxadiazole Hybrids as Anti-Tuberculosis Agents. Bioorg. Med. Chem. Lett. 2016, 26, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Mandewale, M.C.; Thorat, B.; Nivid, Y.; Jadhav, R.; Nagarsekar, A.; Yamgar, R. Synthesis, Structural Studies and Antituberculosis Evaluation of New Hydrazone Derivatives of Quinoline and Their Zn(II) Complexes. J. Saudi Chem. Soc. 2018, 22, 218–228. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through P53-Dependent and -Independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibáñez-Escribano, A.; Fonseca-Berzal, C.; Martínez-Montiel, M.; Álvarez-Márquez, M.; Gómez-Núñez, M.; Lacueva-Arnedo, M.; Espinosa-Buitrago, T.; Martín-Pérez, T.; Escario, J.A.; Merino-Montiel, P.; et al. Thio- and Selenosemicarbazones as Antiprotozoal Agents against Trypanosoma Cruzi and Trichomonas Vaginalis. J. Enzym. Inhib. Med. Chem. 2022, 37, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Ghaffari, R.; Sardari, S.; Farahani, Y.; Mohebbi, S. Discovery of Novel Isatin-Based Thiosemicarbazones: Synthesis, Antibacterial, Antifungal, and Antimycobacterial Screening. Res. Pharm. Sci. 2020, 15, 281. [Google Scholar] [CrossRef]
- Souza, M.R.P.; Coelho, N.P.; Baldin, V.P.; Scodro, R.B.L.; Cardoso, R.F.; da Silva, C.C.; Vandresen, F. Synthesis of Novel (-)-Camphene-Based Thiosemicarbazones and Evaluation of Anti- Mycobacterium Tuberculosis Activity. Nat. Prod. Res. 2019, 33, 3372–3377. [Google Scholar] [CrossRef]
- Beteck, R.M.; Seldon, R.; Jordaan, A.; Warner, D.F.; Hoppe, H.C.; Laming, D.; Khanye, S.D. New Quinolone-Based Thiosemicarbazones Showing Activity Against Plasmodium Falciparum and Mycobacterium Tuberculosis. Molecules 2019, 24, 1740. [Google Scholar] [CrossRef] [Green Version]
- Volynets, G.P.; Tukalo, M.A.; Bdzhola, V.G.; Derkach, N.M.; Gumeniuk, M.I.; Tarnavskiy, S.S.; Starosyla, S.A.; Yarmoluk, S.M. Benzaldehyde Thiosemicarbazone Derivatives against Replicating and Nonreplicating Mycobacterium Tuberculosis. J. Antibiot. (Tokyo) 2019, 72, 218–224. [Google Scholar] [CrossRef]
- Sens, L.; de Souza, A.C.A.; Pacheco, L.A.; Menegatti, A.C.O.; Mori, M.; Mascarello, A.; Nunes, R.J.; Terenzi, H. Synthetic Thiosemicarbazones as a New Class of Mycobacterium Tuberculosis Protein Tyrosine Phosphatase A Inhibitors. Bioorg. Med. Chem. 2018, 26, 5742–5750. [Google Scholar] [CrossRef] [PubMed]
- Marvadi, S.K.; Krishna, V.S.; Surineni, G.; Srilakshmi Reshma, R.; Sridhar, B.; Sriram, D.; Kantevari, S. Synthesis, in Vitro, and in Vivo (Zebra Fish) Antitubercular Activity of 7,8-Dihydroquinolin-5(6H)-Ylidenehydrazinecarbothioamides. Bioorganic Chem. 2020, 96, 103626. [Google Scholar] [CrossRef] [PubMed]
- Aziz, H.; Saeed, A.; Khan, M.A.; Afridi, S.; Jabeen, F.; Hashim, M. Novel N -Acyl-1 H -imidazole-1-carbothioamides: Design, Synthesis, Biological and Computational Studies. Chem. Biodivers. 2020, 17, e1900509. [Google Scholar] [CrossRef] [PubMed]
- Salve, P.S.; Alegaon, S.G. Synthesis of New 7-Chloro-4-Phenoxyquinoline Analogues as Potential Antitubercular Agents. Med. Chem. Res. 2018, 27, 1–14. [Google Scholar] [CrossRef]
- Matsa, R.; Makam, P.; Kaushik, M.; Hoti, S.L.; Kannan, T. Thiosemicarbazone Derivatives: Design, Synthesis and in Vitro Antimalarial Activity Studies. Eur. J. Pharm. Sci. 2019, 137, 104986. [Google Scholar] [CrossRef] [PubMed]
- Shaik, A.B.; Bhandare, R.R.; Nissankararao, S.; Edis, Z.; Tangirala, N.R.; Shahanaaz, S.; Rahman, M.M. Design, Facile Synthesis and Characterization of Dichloro Substituted Chalcones and Dihydropyrazole Derivatives for Their Antifungal, Antitubercular and Antiproliferative Activities. Molecules 2020, 25, 3188. [Google Scholar] [CrossRef]
- Hosny, N.M.; Hassan, N.Y.; Mahmoud, H.M.; Abdel-Rhman, M.H. Synthesis, Characterization and Cytotoxicity of New 2-isonicotinoyl-N-phenylhydrazine-1-carbothioamide and Its Metal Complexes. Appl. Organomet. Chem. 2019, 33, e4998. [Google Scholar] [CrossRef]
- Elsaman, T.; Mohamed, M.S.; Mohamed, M.A. Current Development of 5-Nitrofuran-2-Yl Derivatives as Antitubercular Agents. Bioorganic Chem. 2019, 88, 102969. [Google Scholar] [CrossRef]
- Cabrera, N.; Mora, J.R.; Márquez, E.; Flores-Morales, V.; Calle, L.; Cortés, E. QSAR and Molecular Docking Modelling of Anti-Leishmanial Activities of Organic Selenium and Tellurium Compounds. SAR QSAR Environ. Res. 2021, 32, 29–50. [Google Scholar] [CrossRef]
- Kar, S.; Roy, K.; Leszczynski, J. Applicability Domain: A Step Toward Confident Predictions and Decidability for QSAR Modeling. In Computational Toxicology; Humana Press: New York, NY, USA, 2018. [Google Scholar]
- Almi, I.; Belaidi, S.; Zerroug, E.; Alloui, M.; Ben Said, R.; Linguerri, R.; Hochlaf, M. QSAR Investigations and Structure-Based Virtual Screening on a Series of Nitrobenzoxadiazole Derivatives Targeting Human Glutathione-S-Transferases. J. Mol. Struct. 2020, 1211, 128015. [Google Scholar] [CrossRef]
- Hammoudi, N.-E.-H.; Sobhi, W.; Attoui, A.; Lemaoui, T.; Erto, A.; Benguerba, Y. In Silico Drug Discovery of Acetylcholinesterase and Butyrylcholinesterase Enzymes Inhibitors Based on Quantitative Structure-Activity Relationship (QSAR) and Drug-Likeness Evaluation. J. Mol. Struct. 2021, 1229, 129845. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Predictive QSAR Modeling Based on Diversity Sampling of Experimental Datasets for the Training and Test Set Selection. J. Comput. Aided Mol. Des. 2002, 16, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P. Cheminformatics Analysis of Organic Substituents: Identification of the Most Common Substituents, Calculation of Substituent Properties, and Automatic Identification of Drug-like Bioisosteric Groups. J. Chem. Inf. Comput. Sci. 2003, 43, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Cortes, E.; Mora, J.; Márquez, E. Modelling the Anti-Methicillin-Resistant Staphylococcus Aureus (MRSA) Activity of Cannabinoids: A QSAR and Docking Study. Crystals 2020, 10, 692. [Google Scholar] [CrossRef]
- Cvetković, J.P.; Božić, B.Đ.; Banjac, N.R.; Petrović, J.; Soković, M.; Vitnik, V.D.; Vitnik, Ž.J.; Ušćumlić, G.S.; Valentić, N.V. Synthesis, Antimicrobial Activity and Quantum Chemical Investigation of Novel Succinimide Derivatives. J. Mol. Struct. 2019, 1181, 148–156. [Google Scholar] [CrossRef]
- Eryılmaz, S.; Türk Çelikoğlu, E.; İdil, Ö.; İnkaya, E.; Kozak, Z.; Mısır, E.; Gül, M. Derivatives of Pyridine and Thiazole Hybrid: Synthesis, DFT, Biological Evaluation via Antimicrobial and DNA Cleavage Activity. Bioorganic Chem. 2020, 95, 103476. [Google Scholar] [CrossRef]
- Uzzaman, M.; Junaid, M.; Uddin, M.N. Evaluation of Anti-Tuberculosis Activity of Some Oxotitanium(IV) Schiff Base Complexes; Molecular Docking, Dynamics Simulation and ADMET Studies. SN Appl. Sci. 2020, 2, 880. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.; Shankar, R. 2-Isoxazolines: A Synthetic and Medicinal Overview. ChemMedChem 2021, 16, 430–447. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, D.; Wang, S.; Chen, H.; Wei, N.; Chen, G. Molecular Insight into the Discrepancy of Antitubercular Activity between 8-Nitro and 8-Cyano Benzothiazinones. Chem. Select 2020, 5, 13775–13779. [Google Scholar] [CrossRef]
- Al-Tamimi, A.-M.S.; Mary, Y.S.; Miniyar, P.B.; Al-Wahaibi, L.H.; El-Emam, A.A.; Armaković, S.; Armaković, S.J. Synthesis, Spectroscopic Analyses, Chemical Reactivity and Molecular Docking Study and Anti-Tubercular Activity of Pyrazine and Condensed Oxadiazole Derivatives. J. Mol. Struct. 2018, 1164, 459–469. [Google Scholar] [CrossRef]
- Pitucha, M.; Karczmarzyk, Z.; Swatko-Ossor, M.; Drozd, M.; Wysocki, W.; Ginalska, G.; Urbanczyk-Lipkowska, Z.; Kowalczuk, D.; Morawiak, M. Synthesis and Structural Study of Some N-Acyl-4-Allylsemicarbazides and the Product of Their Cyclization with a Potential Antimicrobial Activity. J. Mol. Struct. 2020, 1219, 128552. [Google Scholar] [CrossRef]
- Kucuk, C.; Yurdakul, S.; Erdem, B. Spectroscopic Characterization, DFT Calculations, and Microbiological Activity of 5-Iodoindole. J. Mol. Struct. 2022, 1252, 132125. [Google Scholar] [CrossRef]
- Dubey, R.P.; Patel, U.H.; Pandya, S.B.; Chaudhary, K.P.; Socha, B.N. Cadmium Complex of Sulfathiazole Dihydrate with Secondary Ligand Pyridine: Structure, DFT Studies, Hirshfeld Surface Analysis and Antimicrobial Activity. Indian J. Phys. 2021, 95, 33–42. [Google Scholar] [CrossRef]
- Lone, I.H.; Khan, K.Z.; Fozdar, B.I. Synthesis, Physicochemical Properties, Antimicrobial and Antioxidant Studies of Pyrazoline Derivatives Bearing a Pyridyl Moiety. Med. Chem. Res. 2014, 23, 363–369. [Google Scholar] [CrossRef]
- Salim, A.S.; Girgis, A.S.; Basta, A.H.; El-saied, H.; Mohamed, M.A.; Bedair, A.H. Comparative DFT Computational Studies with Experimental Investigations for Novel Synthesized Fluorescent Pyrazoline Derivatives. J. Fluoresc. 2018, 28, 913–931. [Google Scholar] [CrossRef]
- Lougheed, K.E.A.; Taylor, D.L.; Osborne, S.A.; Bryans, J.S.; Buxton, R.S. New Anti-Tuberculosis Agents Amongst Known Drugs. Tuberc. Edinb. Scotl. 2009, 89, 364. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Pawara, R.; Pawara, K.; Ahmed, F.; Shirkhedkar, A.; Surana, S. A Structural Insight of Bedaquiline for the Cardiotoxicity and Hepatotoxicity. Tuberculosis 2019, 117, 79–84. [Google Scholar] [CrossRef]
- Ahmad, I.; Jadhav, H.; Shinde, Y.; Jagtap, V.; Girase, R.; Patel, H. Optimizing Bedaquiline for Cardiotoxicity by Structure Based Virtual Screening, DFT Analysis and Molecular Dynamic Simulation Studies to Identify Selective MDR-TB Inhibitors. Silico Pharmacol. 2021, 9. [Google Scholar] [CrossRef]
- Aldred, K.J.; Blower, T.R.; Kerns, R.J.; Berger, J.M.; Osheroff, N. Fluoroquinolone Interactions with Mycobacterium Tuberculosis Gyrase: Enhancing Drug Activity against Wild-Type and Resistant Gyrase. Proc. Natl. Acad. Sci. USA 2016, 113, E839–E846. [Google Scholar] [CrossRef]
- Sarathy, J.; Blanc, L.; Alvarez-Cabrera, N.; O’Brien, P.; Dias-Freedman, I.; Mina, M.; Zimmerman, M.; Kaya, F.; Ho Liang, H.-P.; Prideaux, B.; et al. Fluoroquinolone Efficacy against Tuberculosis Is Driven by Penetration into Lesions and Activity against Resident Bacterial Populations. Antimicrob. Agents Chemother. 2019, 63, e02516-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antimicrobial and Anti-Tubercular Activity of Quinolone Analogues. Available online: https://scialert.net/abstract/?doi=sciintl.2013.336.349 (accessed on 5 December 2022).
- Hotra, A.; Ragunathan, P.; Ng, P.S.; Seankongsuk, P.; Harikishore, A.; Sarathy, J.P.; Saw, W.-G.; Lakshmanan, U.; Sae-Lao, P.; Kalia, N.P.; et al. Discovery of a Novel Mycobacterial F-ATP Synthase Inhibitor and Its Potency in Combination with Diarylquinolines. Angew. Chem. Int. Ed. 2020, 59, 13295–13304. [Google Scholar] [CrossRef]
- Richter, A.; Rudolph, I.; Möllmann, U.; Voigt, K.; Chung, C.W.; Singh, O.M.P.; Rees, M.; Mendoza-Losana, A.; Bates, R.; Ballell, L.; et al. Novel Insight into the Reaction of Nitro, Nitroso and Hydroxylamino Benzothiazinones and of Benzoxacinones with Mycobacterium Tuberculosis DprE1. Sci. Rep. 2018, 8, 13473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.T.; Blicharska, N.; Shilpi, J.A.; Seidel, V. Investigation of the Anti-TB Potential of Selected Propolis Constituents Using a Molecular Docking Approach. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, M.; Sun, F.; Li, S.; Hse, C.-Y.; Jin, C. Screening, Synthesis, and QSAR Research on Cinnamaldehyde-Amino Acid Schiff Base Compounds as Antibacterial Agents. Molecules 2018, 23, 3027. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, H.C.; Singh, M.; Prakash, O.; Khan, F.; Srivastava, S.K.; Bawankule, D.U. QSAR, ADME and Docking Guided Semi-Synthesis and in Vitro Evaluation of 4-Hydroxy-α-Tetralone Analogs for Anti-Inflammatory Activity. SN Appl. Sci. 2020, 2, 2069. [Google Scholar] [CrossRef]
- Jin, Y.; Fan, S.; Lv, G.; Meng, H.; Sun, Z.; Jiang, W.; Van Lanen, S.G.; Yang, Z. Computer-Aided Drug Design of Capuramycin Analogues as Anti-Tuberculosis Antibiotics by 3D-QSAR and Molecular Docking. Open Chem. 2017, 15, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Saxena, M.; Gaur, S.; Prathipati, P.; Saxena, A.K. Synthesis of Some Substituted Pyrazinopyridoindoles and 3D QSAR Studies along with Related Compounds: Piperazines, Piperidines, Pyrazinoisoquinolines, and Diphenhydramine, and Its Semi-Rigid Analogs as Antihistamines (H1). Bioorg. Med. Chem. 2006, 14, 8249–8258. [Google Scholar] [CrossRef]
- Meth-Cohn, O.; Narine, B.; Tarnowski, B. A Versatile New Synthesis of Quinolines and Related Fused Pyridines, Part 5. The Synthesis of 2-Chloroquinoline-3-Carbaldehydes. J. Chem. Soc. Perkin. 1981, 1, 15250. [Google Scholar] [CrossRef]
- Abonia, R.; Insuasty, D.; Castillo, J.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Cobo, J. Synthesis of Novel Quinoline-2-One Based Chalcones of Potential Anti-Tumor Activity. Eur. J. Med. Chem. 2012, 57, 29–40. [Google Scholar] [CrossRef]
- Brudey, K.; Driscoll, J.R.; Rigouts, L.; Prodinger, W.M.; Gori, A.; Al-Hajoj, S.A.; Allix, C.; Aristimuño, L.; Arora, J.; Baumanis, V.; et al. Mycobacterium Tuberculosis Complex Genetic Diversity: Mining the Fourth International Spoligotyping Database (SpolDB4) for Classification, Population Genetics and Epidemiology. BMC Microbiol. 2006, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glynn, J. Beijing/W Genotype Mycobacterium Tuberculosis and Drug Resistance—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/16704829/ (accessed on 29 June 2022).
- Reynaud, Y.; Millet, J.; Rastogi, N. Genetic Structuration, Demography and Evolutionary History of Mycobacterium Tuberculosis LAM9 Sublineage in the Americas as Two Distinct Subpopulations Revealed by Bayesian Analyses. PloS ONE 2015, 10, e0140911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubillos-Ruiz, A.; Sandoval, A.; Ritacco, V.; López, B.; Robledo, J.; Correa, N.; Hernandez-Neuta, I.; Zambrano, M.M.; Del Portillo, P. Genomic Signatures of the Haarlem Lineage of Mycobacterium Tuberculosis: Implications of Strain Genetic Variation in Drug and Vaccine Development. J. Clin. Microbiol. 2010, 48, 3614–3623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but Verify II: A Practical Guide to Chemogenomics Data Curation. J. Chem. Inf. Model. 2016, 56, 1243–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, But Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef]
- Tropsha, A.; Bajorath, J. Computational Methods for Drug Discovery and Design. J. Med. Chem. 2016, 59, 1. [Google Scholar] [CrossRef] [Green Version]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [Green Version]
- Mandewale, M.C.; Thorat, B.; Shelke, D.; Yamgar, R. Synthesis and Biological Evaluation of New Hydrazone Derivatives of Quinoline and Their Cu(II) and Zn(II) Complexes against Mycobacterium Tuberculosis. Bioinorg. Chem. Appl. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.B.; Darji, D.G.; Patel, K.R.; Rajani, D.P.; Rajani, S.D.; Patel, H.D. Synthesis of Novel Quinoline-thiosemicarbazide Hybrids and Evaluation of Their Biological Activities, Molecular Docking, Molecular Dynamics, Pharmacophore Model Studies, and ADME-Tox Properties. J. Heterocycl. Chem. 2020, 57. [Google Scholar] [CrossRef]
- Salve, P.S.; Alegaon, S.G.; Sriram, D. Three-Component, One-Pot Synthesis of Anthranilamide Schiff Bases Bearing 4-Aminoquinoline Moiety as Mycobacterium Tuberculosis Gyrase Inhibitors. Bioorg. Med. Chem. Lett. 2017, 27. [Google Scholar] [CrossRef]
- Wolf, A.; Shahid, M.; Kasam, V.; Ziegler, W.; Hofmann-Apitius, M. In Silico Drug Discovery Approaches on Grid Computing Infrastructures. Curr. Clin. Pharmacol. 2010, 5, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Insuasty, D.; Vidal, O.; Bernal, A.; Marquez, E.; Guzman, J.; Insuasty, B.; Quiroga, J.; Svetaz, L.; Zacchino, S.; Puerto, G.; et al. Antimicrobial Activity of Quinoline-Based Hydroxyimidazolium Hybrids. Antibiotics 2019, 8, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, K.D.; Adhikari, A.V.; Telkar, S.; Chowdhury, I.H.; Mahmood, R.; Pal, N.K.; Row, G.; Sumesh, E. Design, Synthesis and Docking Studies of New Quinoline-3-Carbohydrazide Derivatives as Antitubercular Agents. Eur. J. Med. Chem. 2011, 46, 5283–5292. [Google Scholar] [CrossRef] [PubMed]
- Yuanita, E.; Dharmayani, N.K.T.; YUlfa, M.; Syahri, J. Quantitative Structure–Activity Relationship (QSAR) and Molecular Docking of Xanthone Derivatives as Anti-Tuberculosis Agents. J. Clin. Tuberc. Mycobact. Dis. 2020, 21, 100203. [Google Scholar] [CrossRef]
- Adeniji, S.E.; Adamu Shallangwa, G.; Ebuka Arthur, D.; Abdullahi, M.; Mahmoud, A.Y.; Haruna, A. Quantum Modelling and Molecular Docking Evaluation of Some Selected Quinoline Derivatives as Anti-Tubercular Agents. Heliyon 2020, 6, e03639. [Google Scholar] [CrossRef] [PubMed]
- Mali, S.N.; Pandey, A.; Thorat, B.R.; Lai, C.-H. Multiple 3D- and 2D-Quantitative Structure–Activity Relationship Models (QSAR), Theoretical Study and Molecular Modeling to Identify Structural Requirements of Imidazopyridine Analogues as Anti-Infective Agents against Tuberculosis. Struct. Chem. 2022, 33, 679–694. [Google Scholar] [CrossRef]
- Aher, R.B.; Sarkar, D. 2D-QSAR Modeling and Two-Fold Classification of 1,2,4-Triazole Derivatives for Antitubercular Potency against the Dormant Stage of Mycobacterium Tuberculosis. Mol. Divers. 2022, 26, 1227–1242. [Google Scholar] [CrossRef] [PubMed]
- Nayyar, A.; Malde, A.; Coutinho, E.; Jain, R. Synthesis, Anti-Tuberculosis Activity, and 3D-QSAR Study of Ring-Substituted-2/4-Quinolinecarbaldehyde Derivatives. Bioorg. Med. Chem. 2006, 14, 7302–7310. [Google Scholar] [CrossRef]
- Hosseini, S.; Ketabi, S.; Hasheminasab, G. QSAR Study of Antituberculosis Activity of Oxadiazole Derivatives Using DFT Calculations. J. Recept. Signal Transduct. 2022, 42, 503–511. [Google Scholar] [CrossRef]
- Alam, S.; Khan, F. 3D-QSAR Studies on Maslinic Acid Analogs for Anticancer Activity against Breast Cancer Cell Line MCF-7. Sci. Rep. 2017, 7, 6019. [Google Scholar] [CrossRef]
- Abdel-Aziz, H.; Eldehna, W.; Fares, M.; Al-Rashood, S.; Al-Rashood, K.; Abdel-Aziz, M.; Soliman, D. Synthesis, Biological Evaluation and 2D-QSAR Study of Halophenyl Bis-Hydrazones as Antimicrobial and Antitubercular Agents. Int. J. Mol. Sci. 2015, 16, 8719–8743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminformatics 2012, 4, 2946–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian Inc.: Wallingford Center, CT, USA, 2009. [Google Scholar]
- Mahmud, A.W.; Shallangwa, G.A.; Uzairu, A. QSAR and Molecular Docking Studies of 1,3-Dioxoisoindoline-4-Aminoquinolines as Potent Antiplasmodium Hybrid Compounds. Heliyon 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Oyewole, R.O.; Oyebamiji, A.K.; Semire, B. Theoretical Calculations of Molecular Descriptors for Anticancer Activities of 1, 2, 3-Triazole-Pyrimidine Derivatives against Gastric Cancer Cell Line (MGC-803): DFT, QSAR and Docking Approaches. Heliyon 2020, 6. [Google Scholar] [CrossRef]
- Appell, M.; Tu, Y.-S.; Compton, D.L.; Evans, K.O.; Wang, L.C. Quantitative Structure-Activity Relationship Study for Prediction of Antifungal Properties of Phenolic Compounds. Struct. Chem. 2020, 31, 1621–1630. [Google Scholar] [CrossRef]
- Adeniji, S.E.; Arthur, D.E.; Oluwaseye, A. Computational Modeling of 4-Phenoxynicotinamide and 4-Phenoxypyrimidine-5-Carboxamide Derivatives as Potent Anti-Diabetic Agent against TGR5 Receptor. J. King Saud Univ. Sci. 2020, 32, 102–115. [Google Scholar] [CrossRef]
- Saad, F.A.; El-Metwaly, N.M.; Farghaly, T.A.; Elghalban, M.G.; Shah, R.K.; Al-Hazmi, G.A.; Saleh, K.A.; Alfaifi, M.Y. Illustration for Series of New Metal Ion Complexes Extracted from Pyrazolone Derivative, Spectral, Thermal, QSAR, DFT/B3LYP, Docking and Antitumor Investigations. J. Mol. Liq. 2017, 229. [Google Scholar] [CrossRef]
- Djeradi, H.; Rahmouni, A.; Cheriti, A. Antioxidant Activity of Flavonoids: A QSAR Modeling Using Fukui Indices Descriptors. J. Mol. Model. 2014, 20, 614–627. [Google Scholar] [CrossRef]
- Horst, B. Molecular Descriptors and the Electronic Structure. In Statistical Modelling of Molecular Descriptors in QSAR/QSPR.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. [Google Scholar]
- Naef, R.; Acree, W. Calculation of Five Thermodynamic Molecular Descriptors by Means of a General Computer Algorithm Based on the Group-Additivity Method: Standard Enthalpies of Vaporization, Sublimation and Solvation, and Entropy of Fusion of Ordinary Organic Molecules and Total Phase-Change Entropy of Liquid Crystals. Molecules 2017, 22, 1059. [Google Scholar] [CrossRef] [Green Version]
- Yoshimori, A. Prediction of Molecular Properties Using Molecular Topographic Map. Molecules 2021, 26, 4475. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Zanni, R.; Galvez-Llompart, M.; Garcia-Domenech, R.; Galvez, J. What Place Does Molecular Topology Have in Today’s Drug Discovery? Expert Opin. Drug Discov. 2020, 15, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- García-Jacas, C.R.; Marrero-Ponce, Y.; Vivas-Reyes, R.; Suárez-Lezcano, J.; Martinez-Rios, F.; Terán, J.E.; Aguilera-Mendoza, L. Distributed and Multicore QuBiLS-MIDAS Software v2.0: Computing Chiral, Fuzzy, Weighted and Truncated Geometrical Molecular Descriptors Based on Tensor Algebra. J. Comput. Chem. 2020, 41, 1209–1227. [Google Scholar] [CrossRef] [PubMed]
- Heinze, G.; Wallisch, C.; Dunkler, D. Variable Selection—A Review and Recommendations for the Practicing Statistician. Biom. J. 2018, 60. [Google Scholar] [CrossRef] [Green Version]
- Gramatica, P. Principles of QSAR Models Validation: Internal and External. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Cramer, R.D.; Bunce, J.D.; Patterson, D.E.; Frank, I.E. Crossvalidation, Bootstrapping, and Partial Least Squares Compared with Multiple Regression in Conventional QSAR Studies. Quant. Struct.-Act. Relatsh. 1988, 7, 18–25. [Google Scholar] [CrossRef]
- Rücker, C.; Rücker, G.; Meringer, M. Y-Randomization and Its Variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of Q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational Protein–Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Pradeep Kumar, C.B.; Prathibha, B.S.; Prasad, K.N.N.; Raghu, M.S.; Prashanth, M.K.; Jayanna, B.K.; Alharthi, F.A.; Chandrasekhar, S.; Revanasiddappa, H.D.; Yogesh Kumar, K. Click Synthesis of 1,2,3-Triazole Based Imidazoles: Antitubercular Evaluation, Molecular Docking and HSA Binding Studies. Bioorg. Med. Chem. Lett. 2021, 36, 127810. [Google Scholar] [CrossRef]
- Danne, A.B.; Choudhari, A.S.; Chakraborty, S.; Sarkar, D.; Khedkar, V.M.; Shingate, B.B. Triazole–Diindolylmethane Conjugates as New Antitubercular Agents: Synthesis, Bioevaluation, and Molecular Docking. MedChemComm 2018, 9, 1114–1130. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Farooq, U.; Khan, S.; Sarwar, R.; Mabkhot, Y.N.; Saeed, M.; Alsayari, A.; Muhsinah, A.; Bin Ul-Haq, Z. Pharmacophore Model-Based Virtual Screening, Docking, Biological Evaluation and Molecular Dynamics Simulations for Inhibitors Discovery against α-Tryptophan Synthase from Mycobacterium Tuberculosis. J. Biomol. Struct. Dyn. 2020, 39, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Jena, P.K.; Pradhan, S.K. Arabinosyltransferase C Enzyme of Mycobacterium Tuberculosis, a Potential Drug Target: An Insight from Molecular Docking Study. Heliyon 2020, 6, e02693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khedr, M.A.; Pillay, M.; Chandrashekharappa, S.; Chopra, D.; Aldhubiab, B.E.; Attimarad, M.; Alwassil, O.I.; Mlisana, K.; Odhav, B.; Venugopala, K.N. Molecular Modeling Studies and Anti-TB Activity of Trisubstituted Indolizine Analogues; Molecular Docking and Dynamic Inputs. J. Biomol. Struct. Dyn. 2017, 36, 2163–2178. [Google Scholar] [CrossRef]
- Holas, O.; Ondrejcek, P.; Dolezal, M. Mycobacterium Tuberculosis Enoyl-Acyl Carrier Protein Reductase Inhibitors as Potential Antituberculotics: Development in the Past Decade. J. Enzym. Inhib. Med. Chem. 2015, 30, 629–648. [Google Scholar] [CrossRef]
- Mahapatra, S.; Woolhiser, L.K.; Lenaerts, A.J.; Johnson, J.L.; Eisenach, K.D.; Joloba, M.L.; Boom, W.H.; Belisle, J.T. A Novel Metabolite of Antituberculosis Therapy Demonstrates Host Activation of Isoniazid and Formation of the Isoniazid-NAD + Adduct. Antimicrob. Agents Chemother. 2012, 56, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Chikhale, R.V.; Barmade, M.A.; Murumkar, P.R.; Yadav, M.R. Overview of the Development of DprE1 Inhibitors for Combating the Menace of Tuberculosis. J. Med. Chem. 2018, 61, 8563–8593. [Google Scholar] [CrossRef]
- Oleg, T.; Arthur, J.O. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-Mmpbsa -A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins Struct. Funct. Bioinforma. 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carloni, P.; Rothlisberger, U.; Parrinello, M. The Role and Perspective of Ab Initio Molecular Dynamics in the Study of Biological Systems. Acc. Chem. Res. 2002, 35, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.S.; Kubař, T.; Cui, Q.; Elstner, M. Semiempirical Quantum Mechanical Methods for Noncovalent Interactions for Chemical and Biochemical Applications. Chem. Rev. 2016, 116, 5301–5337. [Google Scholar] [CrossRef] [PubMed]
- Faver, J.; Merz, K.M. The Utility of the HSAB Principle via the Fukui Function in Biological Systems. J. Chem. Theory Comput. 2010, 6, 548–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Wang, Y.; Zhu, W. Nonbonding Interactions of Organic Halogens in Biological Systems: Implications for Drug Discovery and Biomolecular Design. Phys. Chem. Chem. Phys. 2010, 12, 4543–4551. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Santos, A.; Chaves, E.J.F.; Grillo, I.B.; de Freitas, A.S.; Araújo, D.A.M.; Rocha, G.B. Thermochemical and Quantum Descriptor Calculations for Gaining Insight into Ricin Toxin A (RTA) Inhibitors. ACS Omega 2021, 6, 8764–8777. [Google Scholar] [CrossRef]
- Sapse, A.-M. (Ed.) Molecular Orbital Calculations for Biological Systems; Topics in Physical Chemistry; Oxford University Press: New York, NY, USA, 1998; ISBN 978-0-19-509873-0. [Google Scholar]
- Stachowicz, J.; Krajewska-Kulak, E.; Lukaszuk, C.; Niewiadomy, A. Relationship between Antifungal Activity against Candida Albicans and Electron Parameters of Selected N-Heterocyclic Thioamides. Indian J. Pharm. Sci. 2014, 76, 287. [Google Scholar]
- Tanaka, S.; Mochizuki, Y.; Komeiji, Y.; Okiyama, Y.; Fukuzawa, K. Electron-Correlated Fragment-Molecular-Orbital Calculations for Biomolecular and Nano Systems. Phys. Chem. Chem. Phys. 2014, 16, 10310–10344. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. How Well Can New-Generation Density Functional Methods Describe Stacking Interactions in Biological Systems? Phys. Chem. Chem. Phys. 2005, 7, 2701–2705. [Google Scholar] [CrossRef]
- Suarez, M.; Valencia, J.; Cadena, C.; Maiti, R.; Datta, C.; Puerto, G.; Isaza, J.; San Juan, H.; Nagaraja, V.; Guzman, J. Diarylethenes Display In Vitro Anti-TB Activity and Are Efficient Hits Targeting the Mycobacterium Tuberculosis HU Protein. Molecules 2017, 22, 1245. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attribute | 8a | 24a | χ |
|---|---|---|---|
| 8a | 1 | 0.02 | −0.51 |
| 24a | 0.02 | 1 | 0.25 |
| χ | −0.51 | 0.25 | 1 |
| Size | r2 | QLOO | QLMO | Qext | Qboot | a (R2) | a (Q2) | F | |
|---|---|---|---|---|---|---|---|---|---|
| 3 | 0.83 | 0.78 | 0.72 | 0.81 | 0.716 | 0.13 | −0.27 | 47.96 | |
| pMIC = 6.401 + 2.877(8a) − 0.228(24a) − 19.793χ | |||||||||
| Leave-One-Out Validation | External Validation | |||
|---|---|---|---|---|
| Criterion | Result | Assessment | Result | Experimental |
| r2 > 0.6 | 0.83 | PASS | 0.83 | PASS |
| r2val > 0.5 | 0.78 | PASS | 0.81 | PASS |
| (r2val − r20)/r2val < 0.1 | 0.00 | PASS | 0.00 | PASS |
| (r2val − r20)/r2val < 0.1 | 0.06 | PASS | 0.00 | PASS |
| abs (r20 − r′20) < 0.1 | 0.05 | PASS | 0.00 | PASS |
| 0.85 < K < 1.15 | 0.99 | PASS | 0.99 | PASS |
| 0.85 < K′ < 1.15 | 0.99 | PASS | 1.00 | PASS |
| Comp. | R | Yp | MICp (µM) | 8a | 24a | χ | HD | HA | P | MW |
|---|---|---|---|---|---|---|---|---|---|---|
| 11a | H | 5.49 | 0.23 | 0.25 | −2.29 | 0.10 | 3 | 3 | −0.12 | 286.35 |
| 11b | 6-Me | 5.56 | 0.22 | 0.23 | −3.02 | 0.11 | 3 | 3 | 0.97 | 300.38 |
| 11c | 8-Me | 5.93 | 0.10 | 0.54 | −0.93 | 0.11 | 3 | 3 | 0.97 | 300.38 |
| 11d | 7-Cl | 6.82 | 0.14 | 0.57 | −4.26 | 0.11 | 3 | 3 | 1.34 | 320.80 |
| 11e | 6-Br | 7.03 | 0.09 | 0.57 | −4.99 | 0.10 | 3 | 3 | 1.49 | 365.25 |
| Compounds | Vina Score (kcal/mol) | RMS | RMS | |
|---|---|---|---|---|
| DPrE1 | InhA | |||
| 11a | −7.30 | 1.8 | −7.60 | 2.3 |
| 11b | −7.10 | 2.4 | −8.20 | 1.8 |
| 11c | −7.50 | 1.6 | −8.40 | 1.5 |
| 11d | −7.20 | 2.0 | −8.40 | 1.3 |
| 11e | −7.90 | 1.5 | −8.50 | 1.2 |
| BTZ043 | −10.70 | 1.3 | −8.30 | 2.4 |
| INH-NADP complex | −6.6 | 1.5 | −10.40 | 0.8 |
| Enzyme | Compound | van der Waals (kcal/mol) | Electrostatic (kcal/mol) | SASA (kcal/mol) | Binding Energy (kcal/mol) |
|---|---|---|---|---|---|
| InhA | 11c | −38.20 | −7.40 | −4.30 | −19.30 |
| 11d | −36.60 | −77.20 | −4.60 | −71.30 | |
| 11e | −45.0 | −7.30 | −4.20 | −22.90 | |
| DprE1 | 11c | −40.00 | −14.20 | −4.00 | −13.60 |
| 11d | −46.70 | −19.60 | −4.60 | −12.70 | |
| 11e | −38.20 | −13.10 | −3.90 | −14.80 |
 | |||
|---|---|---|---|
| Compound | R | Yield | Time (h) |
| 11a | H | 80 | 6 |
| 11b | 6-CH3 | 70 | 6 |
| 11c | 8-CH3 | 75 | 6 |
| 11d | 7-Cl | 85 | 8 |
| 11e | 6-Br | 85 | 8 |
| Compound | M. bovis BCG | M. tuberculosis H37Rv |
|---|---|---|
| MICs in µM | ||
| 11a | 0.17 | 0.17 |
| 11b | 0.16 | 0.16 |
| 11c | 0.33 | 0.16 |
| 11d | 0.03 | 0.15 |
| 11e | 0.13 | 0.13 |
| Isoniazid | 0.36 | 0.36 |
| Comp. | M. tuberculosis Orphan | M. tuberculosis Beijing | M. tuberculosis LAM 9 | M. tuberculosis Haarlem | M. tuberculosis ATCC 35838 | M. tuberculosis ATCC 35822 |
|---|---|---|---|---|---|---|
| MICs in µM | ||||||
| 11a | 3.49 | 1.75 | 0.35 | 17.46 | 3.49 | 3.49 |
| 11b | 33.29 | 1.66 | 16.65 | 3.33 | 3.33 | 3.33 |
| 11c | 66.58 | 0.33 | 3.29 | 33.29 | 1.66 | 0.33 |
| 11d | 6.23 | 0.33 | 3.12 | 1.56 | 3.33 | 6.23 |
| 11e | 27.38 | 0.27 | 2.74 | 27.37 | 2.73 | 1.37 |
| Oxafloxacin | 2.67 | 2.76 | 2.76 | 2.76 | 2.76 | 2.76 |
| Compound | Cytotoxicity IC50-Vero Cells µM | a | b | c | d | e | f | g | h |
|---|---|---|---|---|---|---|---|---|---|
| Selectivity Index (SI) (SI = IC50/MIC) | |||||||||
| 11a | 1.91 | 11.24 | 11.24 | 0.55 | 1.09 | 5.46 | 0.11 | 0.55 | 0.55 |
| 11b | 3.53 | 22.06 | 22.06 | 0.11 | 2.13 | 0.21 | 1.06 | 1.06 | 1.06 |
| 11c | 1.90 | 5.75 | 11.88 | 0.03 | 5.76 | 0.58 | 0.06 | 1.14 | 5.76 |
| 11d | 1.65 | 55.00 | 11.00 | 0.26 | 5.00 | 0.53 | 1.06 | 0.50 | 0.26 |
| 11e | 2.20 | 16.92 | 16.92 | 0.08 | 8.15 | 0.80 | 0.08 | 0.81 | 1.61 |
| Protein | PDB:ID | Active Site (x, y, z) | Inhibitor | Grilla Box Size (x, y, z) |
|---|---|---|---|---|
| DprE1 | 6HFW | 13.869, −21.448, 37.131 | 8-(oxidanylamino)-2-piperidin-1-yl-6-(trifluoromethyl)-1,3-benzothiazin-4-one (BTZ043) | 22, 22, 22 |
| InhA | 2PR2 | −1.702, −27.226, 15.656 | INH-NADP | 22, 22, 22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valencia, J.; Rubio, V.; Puerto, G.; Vasquez, L.; Bernal, A.; Mora, J.R.; Cuesta, S.A.; Paz, J.L.; Insuasty, B.; Abonia, R.; et al. QSAR Studies, Molecular Docking, Molecular Dynamics, Synthesis, and Biological Evaluation of Novel Quinolinone-Based Thiosemicarbazones against Mycobacterium tuberculosis. Antibiotics 2023, 12, 61. https://doi.org/10.3390/antibiotics12010061
Valencia J, Rubio V, Puerto G, Vasquez L, Bernal A, Mora JR, Cuesta SA, Paz JL, Insuasty B, Abonia R, et al. QSAR Studies, Molecular Docking, Molecular Dynamics, Synthesis, and Biological Evaluation of Novel Quinolinone-Based Thiosemicarbazones against Mycobacterium tuberculosis. Antibiotics. 2023; 12(1):61. https://doi.org/10.3390/antibiotics12010061
Chicago/Turabian StyleValencia, Jhesua, Vivian Rubio, Gloria Puerto, Luisa Vasquez, Anthony Bernal, José R. Mora, Sebastian A. Cuesta, José Luis Paz, Braulio Insuasty, Rodrigo Abonia, and et al. 2023. "QSAR Studies, Molecular Docking, Molecular Dynamics, Synthesis, and Biological Evaluation of Novel Quinolinone-Based Thiosemicarbazones against Mycobacterium tuberculosis" Antibiotics 12, no. 1: 61. https://doi.org/10.3390/antibiotics12010061