
Synthesis, Antibacterial and Antiribosomal Activity of the 3C-Aminoalkyl Modification in the Ribofuranosyl Ring of Apralogs (5-O-Ribofuranosyl Apramycins)
 , , and
, , and
Abstract
:1. Introduction
2. Results
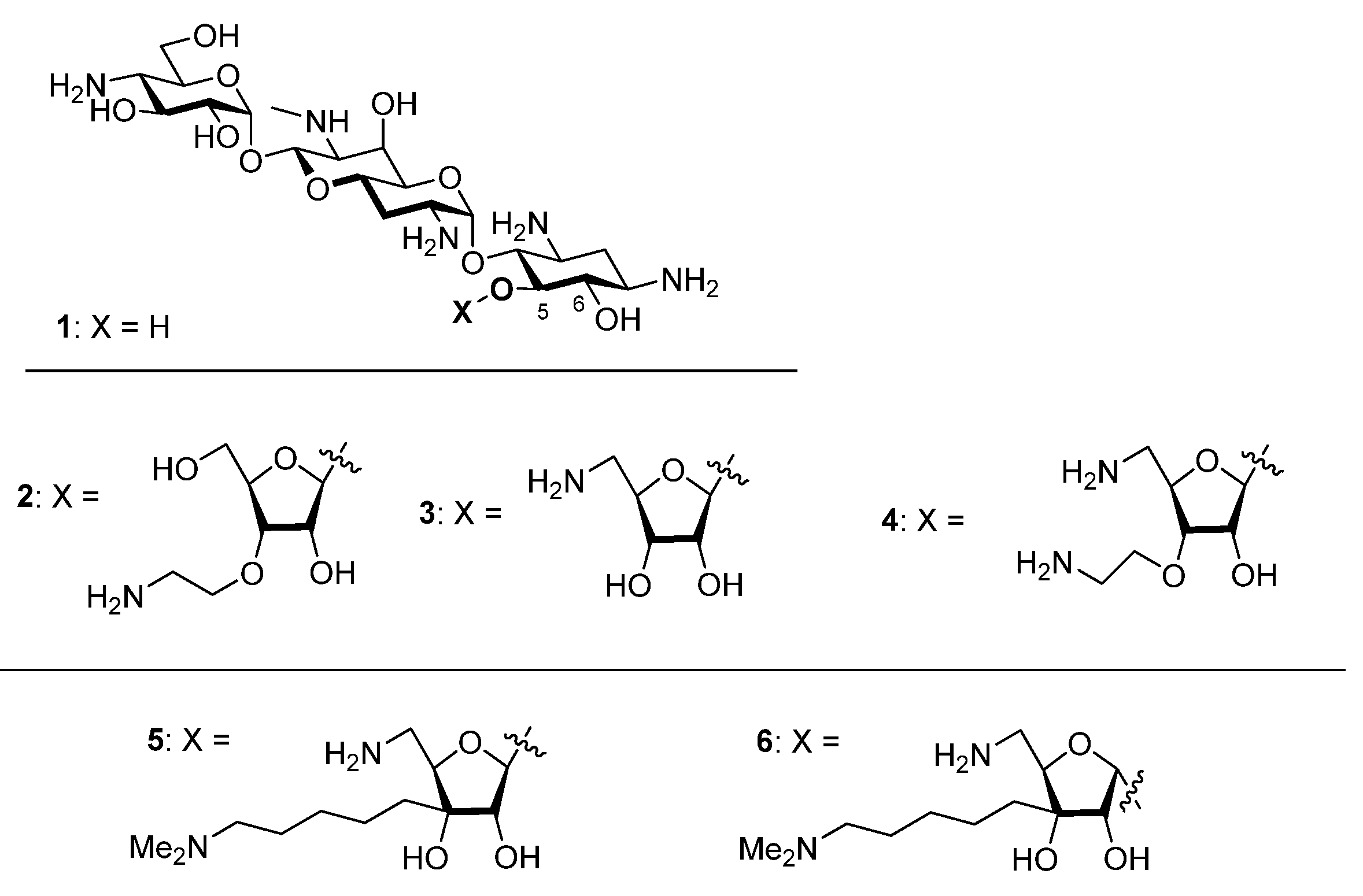
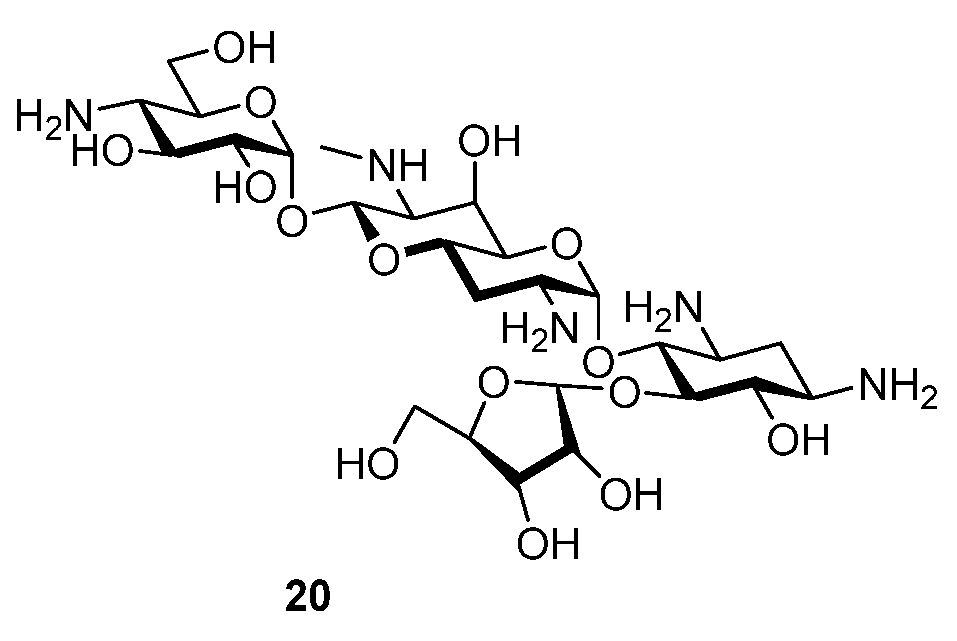
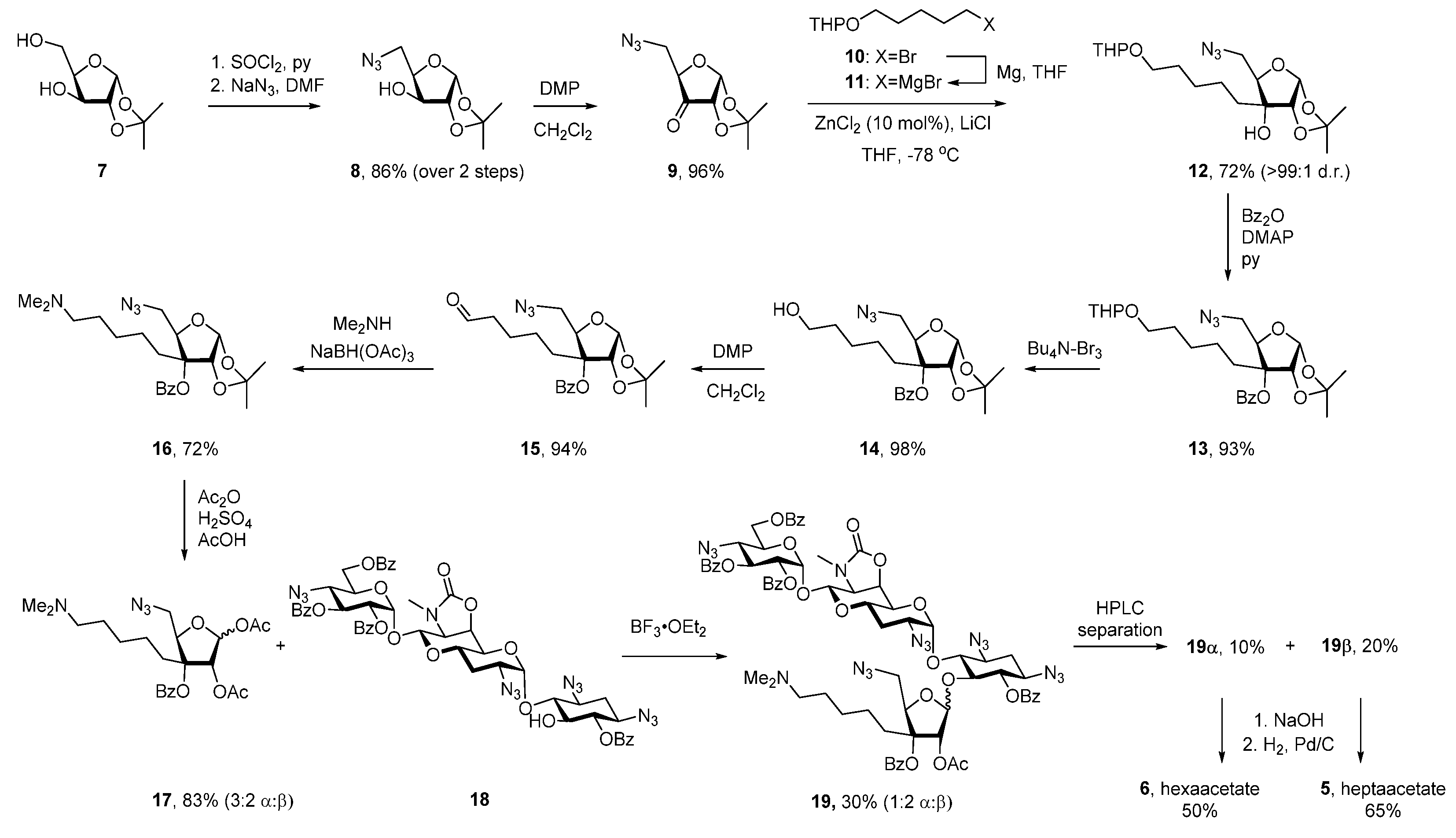
2.1. Synthesis
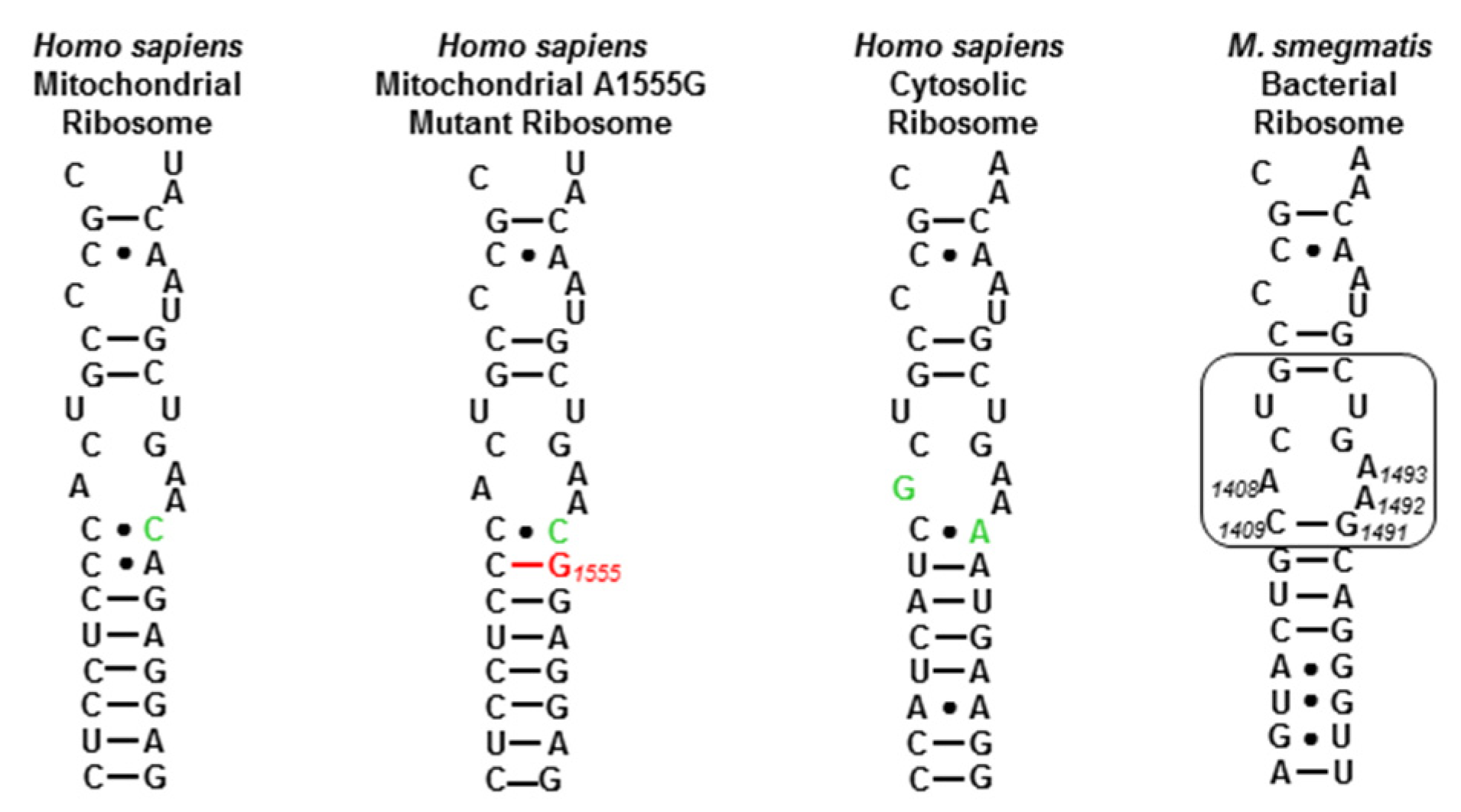
2.2. Activity and Selectivity at the Drug Target
2.3. Antibacterial Activity against Wild-Type Bacterial Strains
2.4. Antibacterial Activity against Resistant Bacterial Strains
2.5. Discussion
3. Conclusions
4. Materials and Methods
4.1. General Experimental
4.2. 5-Azido-5-deoxy-1,2-O-isopropylidene-α-D-xylofuranose (8)
4.3. 5-Azido-5-deoxy-1,2-O-isopropylidene-α-D-erythro-pentofuranos-3-ulose (9)
4.4. 2-(5-Bromopentyloxy)-tetrahydro-2H-pyran (10)
4.5. (5-((Tetrahydro-2H-pyran-2-yl)oxy)pentyl)magnesium bromide (11)
4.6. 5-Azido-5-deoxy-1,2-O-isopropylidene-3-C-(6-(5-((tetrahydro-2H-pyran-2-yl)oxy)pentyl)-α-D-ribofuranose (12)
4.7. 5-Azido-5-deoxy-1,2-O-isopropylidene-3-C-(6-(5-((tetrahydro-2H-pyran-2-yl)oxy)pentyl)-3-O-benzoyl-α-D-ribofuranose (13)
4.8. 5-Azido-5-deoxy-1,2-O-isopropylidene-3-C-(5-hydroxypentyl)-3-O-benzoyl-α-D-ribofuranose (14)
4.9. 5-Azido-5-deoxy-1,2-O-isopropylidene-3-C-(5-oxopentyl)-3-O-benzoyl-α-D-ribofuranose (15)
4.10. 5-Azido-5-deoxy-1,2-O-isopropylidene-3-C-(5-(dimethylamino)pentyl)-3-O-benzoyl-α-D-ribofuranose (16)
4.11. 1,2-Di-O-acetyl-5-azido-5-deoxy-3-C-(5-(dimethylamino)pentyl)-3-O-benzoyl-α/β-D-ribofuranose (17)
4.12. 5-O-[5′′′-Azido-5′′′-deoxy-2′′′-O-acetyl-3-C-(5-(dimethylamino)pentyl)-3-O-benzoyl-α/β-D-ribofuranosyl]-6,2′′,3′′,6′′-tetra-O-benzoyl-1,3,2′,4′′-tetraazido-1,3,2′,4′′-tetra(desamino)-6′,7′-oxazolidino-apramycin trifluoroacetate (19)
4.13. 5-O-[5′′′-Amino-5′′′-deoxy-3-C-(5-(dimethylamino)pentyl)-β-D-ribofuranosyl]-apramycin heptaacetate (5)
4.14. 5-O-[5′′′-Amino-5′′′-deoxy-3-C-(5-(dimethylamino)pentyl)-α-D-ribofuranosyl]-apramycin hexaacetate (6)
4.15. Cell-Free Luciferase Translation Assays
4.16. Antibacterial Inhibition Assays
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, L.; Ye, X.S. Development of Aminoglycoside Antibiotics Effective against Resistant Bacterial Strains. Curr. Top. Med. Chem. 2010, 10, 1898–1926. [Google Scholar] [CrossRef] [PubMed]
- Magnet, S.; Blanchard, J.S. Molecular Insights into Aminoglycoside Action and Resistance. Chem. Rev. 2005, 105, 477–497. [Google Scholar] [CrossRef] [PubMed]
- Garneau-Tsodikova, S.; Labby, K.J. Mechanisms of Resistance to Aminoglycoside Antibiotics: Overview and Perspectives. Med. Chem. Commun. 2016, 7, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Zárate, S.G.; De la Cruz Claure, M.L.; Benito-Arenas, R.; Revuelta, R.; Santana, A.G.; Bastida, A. Overcoming Aminoglycoside Enzymatic Resistance: Design of Novel Antibiotics and Inhibitors. Molecules 2018, 23, 284. [Google Scholar] [CrossRef] [Green Version]
- Matt, T.; Ng, C.L.; Lang, K.; Sha, S.-H.; Akbergenov, R.; Shcherbakov, D.; Meyer, M.; Duscha, S.; Xie, J.; Dubbaka, S.R.; et al. Dissociation of Antibacterial Activity and Aminoglycoside Ototoxicity in the 4-Monosubstituted 2-Deoxystreptamine Apramycin. Proc. Natl. Acad. Sci. USA 2012, 109, 10984–10989. [Google Scholar] [CrossRef] [Green Version]
- Juhas, M.; Widlake, E.; Teo, J.; Huseby, D.L.; Tyrrell, J.M.; Polikanov, Y.; Ercan, O.; Petersson, A.; Cao, S.; Aboklaish, A.F.; et al. In-vitro Activity of Apramycin against Multidrug-, Carbapenem-, and Aminoglycoside-Resistant Enterobacteriaceae and Acinetobacter baumannii. J. Antimicrob. Chemother. 2019, 74, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.; Cao, S.; Nilsson, A.; Erlandsson, M.; Hotop, S.-K.; Kuka, J.; Hansen, J.; Haldimann, K.; Grinberga, S.; Fernández, T.B.; et al. Antibacterial Activity of Apramycin at Acidic pH Warrants Wide Therapeutic Window in the Treatment of Complicated Urinary Tract Infections and Acute Pyelonephritis. EBioMedicine 2021, 73, 103652–103661. [Google Scholar] [CrossRef]
- Becker, K.; Aranzana-Climent, V.; Cao, S.; Nilsson, A.; Shariatgorji, R.; Haldimann, K.; Platzack, B.; Hughes, D.; Andren, P.E.; Böttger, E.C.; et al. Efficacy of EBL-1003 (Apramycin) against Acinetobacter baumannii Lung Infections in Mice. Clin. Microbiol. Infect. 2021, 27, 1315–1321. [Google Scholar] [CrossRef]
- Smith, K.P.; Kirby, J.E. Evaluation of apramycin activity against carbapenem-resistant and -susceptible strains of Enterobacteriaceae. Diagn. Microbiol. Infect. Dis. 2016, 86, 439–441. [Google Scholar] [CrossRef]
- Kang, A.D.; Smith, K.P.; Eliopoulos, G.M.; Berg, A.H.; McCoy, C.; Kirby, J.E. In vitro Apramycin Activity against multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa. Diagn. Microbiol. Infect. Dis. 2017, 88, 188–191. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, L.; Zhang, X.; Feng, Y.; Zong, Z. In vitro activity of neomycin, streptomycin, paromomycin and apramycin against carbapenem-resistant Enterobacteriaceae clinical strains. Front. Microbiol. 2017, 8, 2275–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holbrook, S.Y.L.; Garneau-Tsodikova, S. Evaluation of Aminoglycoside and Carbapenem Resistance in a Collection of Drug-Resistant Pseudomonas aeruginosa Clinical Isolates. Microb. Drug Resist. 2018, 24, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.; Nafplioti, K.; Chatzikonstantinou, M.; Giamarellou, H.; Souli, M. Evaluation of apramycin activity against carbapenem-resistant Enterobacteriaceae and Acinetobacter baumannii. In Proceedings of the 28th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Madrid, Spain, 21–24 April 2018; p. p. 0096.
- Nafplioti, K.; Galani, I.; Adamou, P.; Moraitou, E.; Giannopoulou, P.; Chra, P.; Damala, M.; Vogiatzakis, E.; Trikka-Graphakos, E.; Baka, V.; et al. Epidemic Dissemination of a Carbapenem-Resistant Acinetobacter baumannii Clone Carrying armA. In Proceedings of the 28th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID 2018), Madrid, Spain, 21–24 April 2018; p. p. 1105.
- Kang, A.D.; Smith, K.P.; Berg, A.H.; Truelson, K.A.; Eliopoulos, G.M.; McCoy, C.; Kirby, J.E. Efficacy of Apramycin against Multidrug-Resistant Acinetobacter baumannii in the Murine Neutropenic Thigh Model. Antimicrob. Agent. Chemother. 2018, 62, e02585-17. [Google Scholar] [CrossRef] [Green Version]
- Truelson, K.A.; Brennan-Krohn, T.; Smith, K.P.; Kirby, J.E. Evaluation of apramycin activity against methicillin-resistant, methicillin-sensitive, and vancomycin-intermediate Staphylococcus aureus clinical isolates. Diagn. Microbiol. Infect. Dis. 2018, 92, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Riedel, S.; Vijayakumar, D.; Berg, G.; Kang, A.D.; Smith, K.P.; Kirby, J.E. Evaluation of apramycin against spectinomycin-resistant and -susceptible strains of Neisseria gonorrhoeae. J. Antimicrob. Chemother. 2019, 74, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Brennan-Krohn, T.; Kirby, J.E. Synergistic combinations and repurposed antibiotics active against the pandrug-resistant Klebsiella pneumoniae Nevada strain. Antimicrob. Agent. Chemother. 2019, 63, e01374-19. [Google Scholar] [CrossRef] [Green Version]
- Hao, M.; Shi, X.-M.; Lv, J.; Niu, S.; Cheng, S.; Du, H.; Yu, F.; Tang, Y.-W.; Kreiswirth, B.N.; Zhang, H.; et al. In vitro Activity of Apramycin against Carbapenem-Resistant and Hypervirulaent Klebsiella pneumoniae Isolates. Front. Microbiol. 2020, 11, 425. [Google Scholar] [CrossRef] [Green Version]
- Quirke, J.C.K.; Rajasekaran, P.; Sarpe, V.A.; Sonousi, A.; Osinnii, I.; Gysin, M.; Haldimann, K.; Fang, Q.-J.; Shcherbakov, D.; Hobbie, S.N.; et al. Apralogs: Apramycin 5-O-Glycosides and Ethers with Improved Antibacterial Activity and Ribosomal Selectivity and Reduced Susceptibility to the Aminoacyltranserferase (3)-IV Resistance Determinant. J. Am. Chem. Soc. 2020, 142, 530–544. [Google Scholar] [CrossRef]
- Sonousi, A.; Quirke, J.C.K.; Waduge, P.; Janusic, T.; Gysin, M.; Haldimann, K.; Xu, S.; Hobbie, S.N.; Sha, S.-H.; Schacht, J.; et al. An Advanced Apralog with Increased in-vitro and in-vivo Activity toward Gram-negative Pathogens and Reduced ex-vivo Cochleotoxicity. Chem. Med. Chem. 2021, 16, 335–339. [Google Scholar] [CrossRef]
- Quirke, J.C.K.; Sati, G.C.; Sonousi, A.; Gysin, M.; Haldimann, K.; Böttger, E.C.; Vasella, A.; Hobbie, S.N.; Crich, D. Structure-Activity Relationships for 5′’-Modifications of 4,5-Aminoglycoside Antibiotics. Chem. Med. Chem. 2022, 17, e202200120. [Google Scholar] [CrossRef]
- Abe, Y.; Nakagawa, S.; Naito, T.; Kawaguchi, H. Synthesis and Activity of 6-O-(3-Amino-3-deoxy-α-D-glucopyranosyl)- and 5-O-(β-D-ribofuranosyl)apramycins. J. Antibiot. 1981, 34, 1434–1446. [Google Scholar] [CrossRef] [Green Version]
- Zada, S.L.; Baruch, B.B.; Simhaev, L.; Engel, H.; Fridman, M. Chemical Modifications Reduce Auditory Cell Damage Induced by Aminoglycoside Antibiotics. J. Am. Chem. Soc. 2020, 142, 3077–3087. [Google Scholar] [CrossRef] [PubMed]
- Mandhapati, A.R.; Shcherbakov, D.; Duscha, S.; Vasella, A.; Böttger, E.C.; Crich, D. Importance of the 6′-Hydroxy Group and its Configuration for Apramycin Activity. ChemMedChem 2014, 9, 2074–2083. [Google Scholar] [CrossRef] [PubMed]
- Mandhapati, A.R.; Yang, G.; Kato, T.; Shcherbakov, D.; Hobbie, S.N.; Vasella, A.; Böttger, E.C.; Crich, D. Structure-Based Design and Synthesis of Apramycin-Paromomycin Analogues. Importance of the Configuration at the 6′-Position and Differences Between the 6′-Amino and Hydroxy Series. J. Am. Chem. Soc. 2017, 139, 14611–14619. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.M.; Bols, M. On the Nature of the Electronic Effect of Multiple Hydroxyl Groups in the 6-Membered Ring—The Effects Are Additive But Steric Hindrance Plays A Role Too. Org. Biomol. Chem. 2017, 15, 1164–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansurova, M.; Klusák, V.; Nešněrová, P.; Muck, A.; Doubský, J.; Svatoš, A. Design and synthesis of bombykol analogues for probing pheromone-binding protein–ligand interactions. Front. Physiol. 2009, 65, 1069–1076. [Google Scholar] [CrossRef]
- Christensen, H.M.; Oscarson, S.; Jensen, H.H. Common side reactions of the glycosyl donor in chemical glycosylation. Carbohydr. Res. 2015, 408, 51–95. [Google Scholar] [CrossRef]
- Armstrong, E.S.; Kostrub, C.F.; Cass, R.T.; Moser, H.E.; Serio, A.W.; Miller, G.H. Aminoglycosides. In Antibiotic Discovery and Development; Dougherty, T.J., Pucci, M.J., Eds.; Springer Science+Business Media: New York, NY, USA, 2012; pp. 229–269. [Google Scholar]
- Böttger, E.C.; Crich, D. Aminoglycosides: Time for Resurrection of a Neglected Class of Antibacterials? ACS Infect. Dis. 2020, 6, 168–172. [Google Scholar] [CrossRef]
- Brodersen, D.E.; Clemons, W.M.; Carter, A.P.; Morgan-Warren, R.; Wimberly, B.T.; Ramakrishnan, V. The Structural Basis for the Action of the Antibiotics Tetracycline, Pactamycin, and Hygromycin B on the 30S Ribosomal Subunit. Cell 2000, 103, 1143–1154. [Google Scholar] [CrossRef] [Green Version]
- Moazed, D.; Noller, H.F. Interaction of Antibiotics with Functional Sites in 16S Ribosomal RNA. Nature 1987, 327, 389–394. [Google Scholar] [CrossRef]
- François, B.; Russell, R.J.M.; Murray, J.B.; Aboul-ela, F.; Masquid, B.; Vicens, Q.; Westhof, E. Crystal Structures of Complexes Between Aminoglycosides and Decoding A Site Oligonucleotides: Role of the Number of rings and Positive Charges in the Specific Binding Leading to Miscoding. Nucleic Acids Res. 2005, 33, 5677–5690. [Google Scholar] [CrossRef] [PubMed]
- Fourmy, D.; Recht, M.I.; Blanchard, S.C.; Puglisi, J.D. Structure of the A Site of Escherichia coli 16S Ribosomal RNA Complexed with an Aminoglycoside Antibiotic. Science 1996, 274, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhou, D.; Steitz, T.A.; Polikanov, Y.S.; Gagnon, M.G. Ribosome-Targeting Antibiotics: Modes of Action, Mechanisms of Resistance, and Implications for Drug Design. Ann. Rev. Biochem. 2018, 87, 451–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duscha, S.; Boukari, H.; Shcherbakov, D.; Salian, S.; Silva, S.; Kendall, A.; Kato, T.; Akbergenov, R.; Perez-Fernandez, D.; Bernet, B.; et al. Identification and Evaluation of Improved 4′-O-(Alkyl) 4,5-Disubstituted 2-Deoxystreptamines as Next Generation Aminoglycoside Antibiotics. mBio 2014, 5, e01827-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbie, S.N.; Kalapala, S.K.; Akshay, S.; Bruell, C.; Schmidt, S.; Dabow, S.; Vasella, A.; Sander, P.; Böttger, E.C. Engineering the rRNA Decoding Site of Eukaryotic Cytosolic Ribosomes in Bacteria. Nucl. Acids Res. 2007, 35, 6086–6093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Karasawa, T.; Steyger, P.S. Aminoglycoside-Induced Cochleotoxicity: A Review. Front. Cell. Neurosci. 2017, 11, 308. [Google Scholar] [CrossRef] [Green Version]
- Böttger, E.C.; Schacht, J. The Mitochondrion: A Perpetrator of Acquired Hearing Loss. Hearing Res. 2013, 303, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Prezant, T.R.; Agapian, J.V.; Bohlman, M.C.; Bu, X.; Öztas, S.; Qiu, W.-Q.; Arnos, K.S.; Cortopassi, G.A.; Jaber, L.; Rotter, J.I.; et al. Mitochondrial Ribosomal RNA Mutation Associated with Both Antibiotic-Induced and Non-Syndromic Deafness. Nat. Genet. 1993, 4, 289–294. [Google Scholar] [CrossRef]
- Huth, M.E.; Ricci, A.J.; Cheng, A.G. Mechanisms of Aminoglycoside Ototoxicity and Targets of Hair Cell Protection. Int. J. Otolaryngol. 2011, 2011, 937861–937879. [Google Scholar] [CrossRef] [Green Version]
- Hobbie, S.N.; Akshay, S.; Kalapala, S.K.; Bruell, C.; Shcherbakov, D.; Böttger, E.C. Genetic Analysis of Interactions with Eukaryotic rRNA Identify the Mitoribosome as Target in Aminoglycoside Ototoxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 20888–20893. [Google Scholar] [CrossRef]
- Hobbie, S.N.; Bruell, C.M.; Akshay, S.; Kalapala, S.K.; Shcherbakov, D.; Böttger, E.C. Mitochondrial Deafness Alleles Confer Misreading of the Genetic Code. Proc. Natl. Acad. Sci. USA 2008, 105, 3244–3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Guan, M.-X. Interaction of Aminoglycosides with Human Mitochondrial 12S rRNA Carrying the Deafness-Associated Mutation. Antimicrob. Agent. Chemother. 2009, 53, 4612–4618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, G.D.; Thompson, P.R. Aminoglycoside Phosphotransferases: Proteins, Structure, and Mechanism. Front. Biosci. 1999, 4, d9–d21. [Google Scholar]
- Plattner, M.; Gysin, M.; Haldimann, K.; Becker, K.; Hobbie, S.N. Epidemiologic, Phenotypic, and Structural Characterization of Aminoglycoside-Resistance Gene AAC(3)-IV. Int. J. Mol. Sci. 2020, 21, 6133. [Google Scholar] [CrossRef]
- Livermore, D.M.; Mushtaq, S.; Warner, M.; Zhang, J.-C.; Maharjan, S.; Doumith, M.; Woodford, N. Activity of Aminoglycosides, Including ACHN-490, against Carbapenem-Resistant Enterobacteriaceae Isolates. J. Antimicrob. Chemother. 2011, 66, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Akolkar, N.P.; Adhyapak, J.P.; Aradhye, J.D.; Kumbhani, A.S.; Panchal, B.M.; Jivani, J.K.; Samanta, B.; Pal, R.K.; Thennati, R. Preparation of Amino glycoside Acyl Cyanopyrrolidine Derivatives for Treating or Preventing Diseases Associated with Dipeptidyl Peptidase IV. WO 2009116067 A2. 24 December 2009.
- Ferreira, S.B.; Sodero, A.C.R.; Cardoso, M.F.C.; Lima, E.S.; Kaiser, C.R.; Silva, F.P.; Ferreira, V.F. Synthesis, biological activity, and molecular modeling studies of 1H-1,2,3-triazole derivatives of carbohydrates as α-glucosidases inhibitors. J. Med. Chem. 2010, 53, 2364–2375. [Google Scholar] [CrossRef]
- Ewing, D.F.; Goethals, G.; Mackenzie, G.; Martin, P.; Ronco, G.; Vanbaelinghem, L.; Villa, P. Novel reversed cyclonucleoside analogues with a D-ribofuranose glycone. Carbohydr. Res. 1999, 321, 190–196. [Google Scholar] [CrossRef]
- Lin, H.-S.; Paquette, L.A. A convenient method for determining the concentration of Grignard reagents. Synth. Commun. 1994, 24, 2503–2506. [Google Scholar] [CrossRef]
- Clinical Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 10th ed.; Approved Standard M07-A10; CLSI: Wayne, PA, USA, 2015. [Google Scholar]
- Lubriks, D.; Zogota, R.; Sarpe, V.A.; Matsushita, T.; Sati, G.C.; Haldimann, K.; Gysin, M.; Böttger, E.C.; Vasella, A.; Suna, E.; et al. Synthesis and Antibacterial Activity of Propylamycin Derivatives Functionalized at the 5′- and Other Positions with a View to Overcoming Resistance due to Aminoglycoside Modifying Enzymes. ACS Infect. Dis. 2021, 7, 2413–2424. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antiribosomal Activity | Selectivity | ||||||
|---|---|---|---|---|---|---|---|
| wt | Mit13 | A1555G | Cyt14 | Mit13 | A1555G | Cyt14 | |
| Apramycin 1 | 0.15 | 114 | 105 | 158 | 760 | 700 | 1053 |
| Apralog 2 | 0.071 | 68 | 13 | 190 | 955 | 188 | 2669 |
| Apralog 3 | 0.13 | 121 | 87 | 111 | 909 | 652 | 837 |
| Apralog 4 | 0.031 | 46 | 22 | 51 | 1494 | 699 | 1653 |
| Apralog 5 | 0.077 | 72 | 39 | 116 | 934 | 510 | 1505 |
| Apralog 6 | 0.071 | 62 | 39 | 114 | 871 | 545 | 1599 |
| Species | MRSA | E. coli | K. pneu. | Enterob. | A. baum. | P. aerug.b |
|---|---|---|---|---|---|---|
| Strain | AG038 | ATCC 25922 | AG215 | AG290 | AG309 | AG220 |
| Apramycin 1 | 4 | 4 | 1–2 | 2–4 | 4 | 4 |
| Apralog 2 | 2–4 | 2 | 1–2 | 2 | 8 | 16–32 |
| Apralog 3 | 2 | 4 | 1–2 | 2 | 8 | 4–8 |
| Apralog 4 | 1–2 | 1–2 | 0.5–1 | 1 | 4 | 2 |
| Apralog 5 | 2–4 | 2–4 | 2 | 4 | 16 | 8–16 |
| Apralog 6 | 2–4 | 2–4 | 2 | 2–4 | 16 | 8–16 |
| Resistance det | WT- Parental | APH(3′)-Ia | APH(3′)-IIa | APH(3′)-IIb | APH(3′)-VI | AAC(3)-IV | ArmA | RmtB |
|---|---|---|---|---|---|---|---|---|
| Strain | DH5α | EC122 | EC123 | EC125 | EC127 | EC118 | EC102 | EC103 |
| Apramycin 1 | 1–2 | 0.5–1 | 0.5–1 | 0.5–1 | 0.5–1 | 64 | 1 | 0.5–1 |
| Apralog 2 | 2 | 4–8 | 1–2 | 1 | 1–2 | 2–4 | 2 | 0.5–1 |
| Apralog 4 | 2 | 1–2 | 1 | 1 | 1–2 | 4 | 1–2 | 2 |
| Apralog 5 | 1–2 | 1–2 | 0.5–1 | 1 | 0.5–1 | 4 | 2–4 | 1 |
| Apralog 6 | 1–2 | 2 | 0.5–1 | 1 | 1 | 4 | 2–4 | 1–2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lubriks, D.; Haldimann, K.; Hobbie, S.N.; Vasella, A.; Suna, E.; Crich, D. Synthesis, Antibacterial and Antiribosomal Activity of the 3C-Aminoalkyl Modification in the Ribofuranosyl Ring of Apralogs (5-O-Ribofuranosyl Apramycins). Antibiotics 2023, 12, 25. https://doi.org/10.3390/antibiotics12010025
Lubriks D, Haldimann K, Hobbie SN, Vasella A, Suna E, Crich D. Synthesis, Antibacterial and Antiribosomal Activity of the 3C-Aminoalkyl Modification in the Ribofuranosyl Ring of Apralogs (5-O-Ribofuranosyl Apramycins). Antibiotics. 2023; 12(1):25. https://doi.org/10.3390/antibiotics12010025
Chicago/Turabian StyleLubriks, Dmitrijs, Klara Haldimann, Sven N. Hobbie, Andrea Vasella, Edgars Suna, and David Crich. 2023. "Synthesis, Antibacterial and Antiribosomal Activity of the 3C-Aminoalkyl Modification in the Ribofuranosyl Ring of Apralogs (5-O-Ribofuranosyl Apramycins)" Antibiotics 12, no. 1: 25. https://doi.org/10.3390/antibiotics12010025