
Aminoglycoside-Modifying Enzymes Are Sufficient to Make Pseudomonas aeruginosa Clinically Resistant to Key Antibiotics


Abstract
:1. Introduction
2. Results
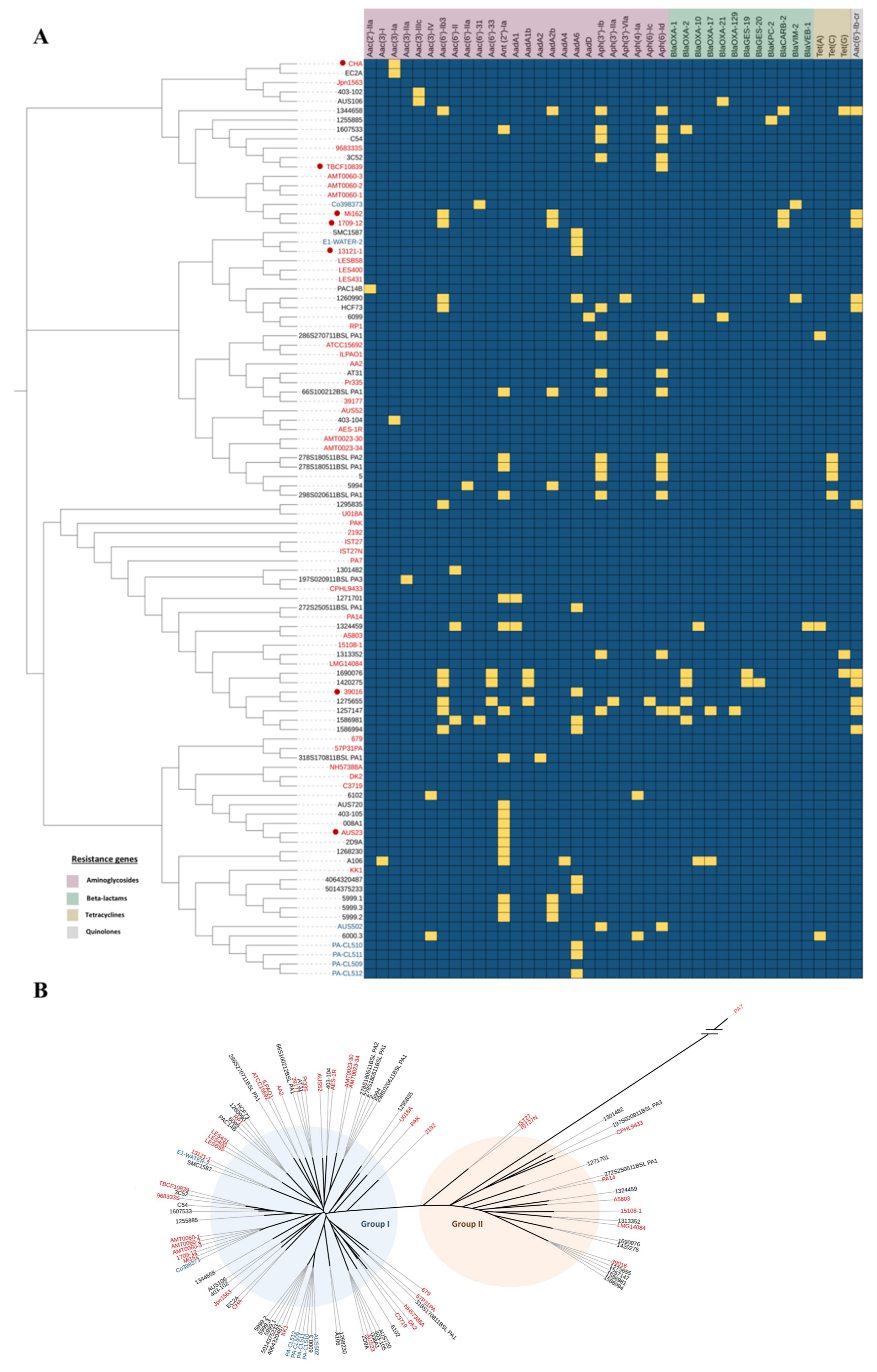
2.1. Identification of Horizontally Transferred Resistance Genes
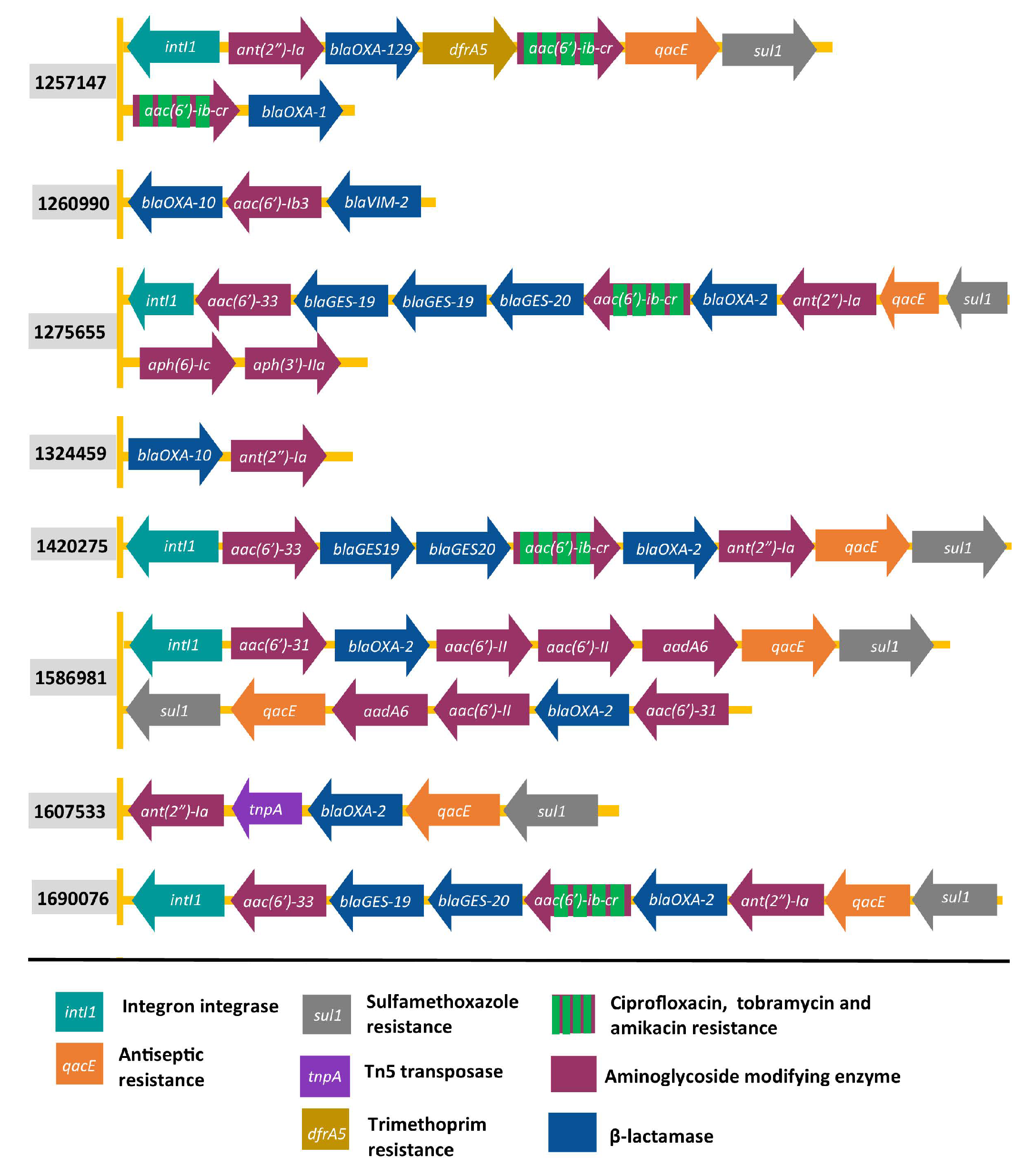
2.2. Organisation of Genes Encoding AMEs
2.3. MICs of Clinical Isolates with Frequent AMEs
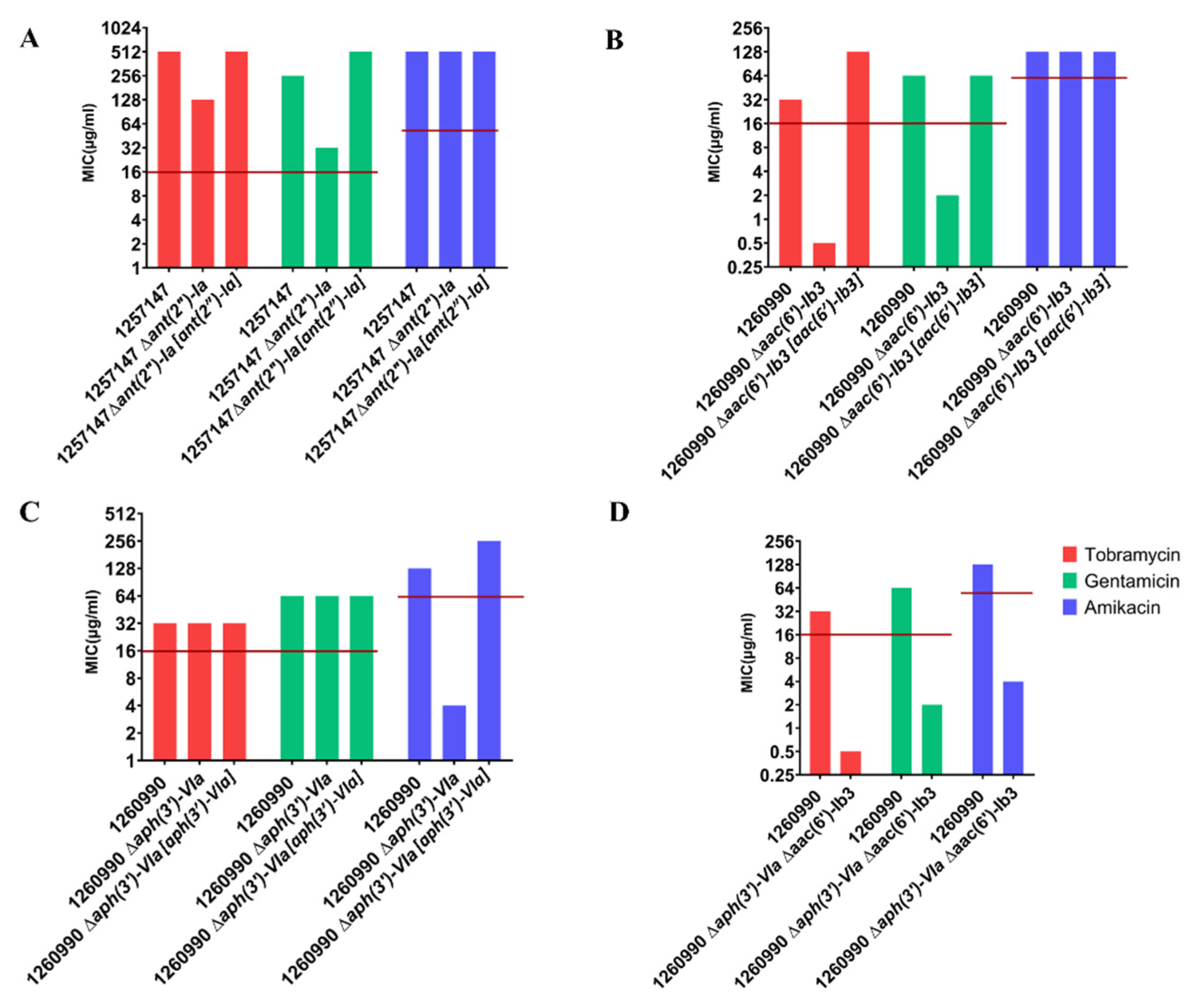
2.4. Deleting AME-Encoding Genes in Clinical Isolates
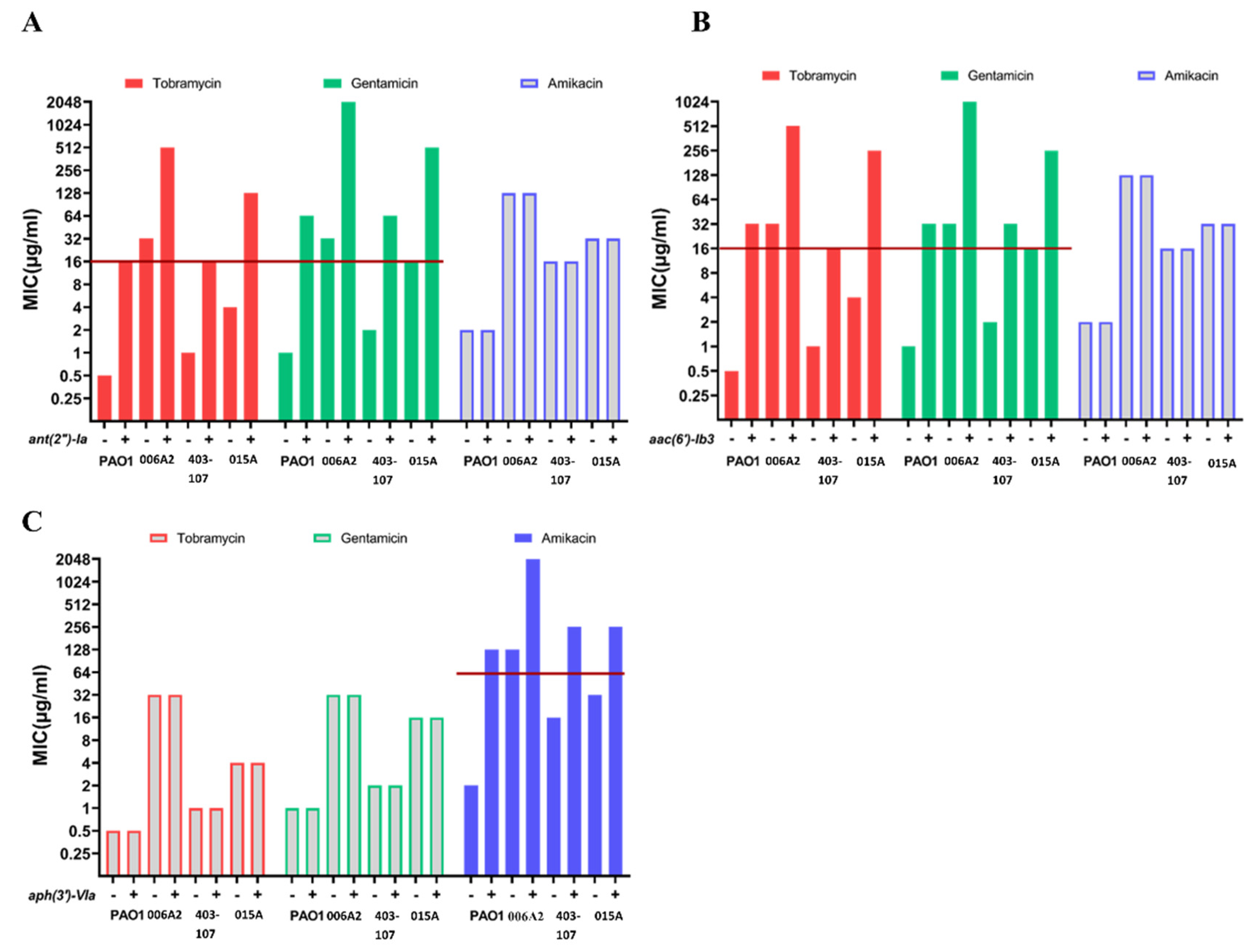
2.5. The Effects of Introduced AMEs in AME-Free P. aeruginosa
2.6. The Effects of AMEs in Combination with Other Resistance Mechanisms
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Minimum Inhibitory Concentration (MIC) Testing
4.3. Genetic Manipulations
4.4. Induced Expression of Aminoglycoside Modifying Enzymes
4.5. Genome Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moradali, M.F.; Ghods, S.; Rehm, B.H. Pseudomonas aeruginosa lifestyle: A paradigm for adaptation, survival, and persistence. Front. Cell. Infect. Microbiol. 2017, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Thankappan, B.; Jayaraman, A.; Gupta, A. Evaluation of Antibiotic Tolerance in Pseudomonas aeruginosa for Aminoglycosides and its Predicted Gene Regulations Through In-silico Transcriptomic Analysis. Microbiol. Res. 2021, 12, 630–645. [Google Scholar] [CrossRef]
- Mantero, M.; Gramegna, A.; Pizzamiglio, G.; D’Adda, A.; Tarsia, P.; Blasi, F. Once daily aerosolised tobramycin in adult patients with cystic fibrosis in the management of Pseudomonas aeruginosa chronic infection. Multidiscip. Respir. Med. 2017, 12, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Ehsan, Z.; Clancy, J.P. Management of Pseudomonas aeruginosa infection in cystic fibrosis patients using inhaled antibiotics with a focus on nebulized liposomal amikacin. Future Microbiol. 2015, 10, 1901–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cag, Y.; Caskurlu, H.; Fan, Y.; Cao, B.; Vahaboglu, H. Resistance mechanisms. Ann. Transl. Med. 2016, 4, 326. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.-J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Poole, K. Aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2005, 49, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Pachori, P.; Gothalwal, R.; Gandhi, P. Emergence of antibiotic resistance Pseudomonas aeruginosa in intensive care unit; a critical review. Genes Dis. 2019, 6, 109–119. [Google Scholar] [CrossRef]
- Singh, M.; Yau, Y.C.; Wang, S.; Waters, V.; Kumar, A. MexXY efflux pump overexpression and aminoglycoside resistance in cystic fibrosis isolates of Pseudomonas aeruginosa from chronic infections. Can. J. Microbiol. 2017, 63, 929–938. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H.; Takatsuka, Y. Mechanisms of RND multidrug efflux pumps. Biochim. Biophys. Acta 2009, 1794, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Prickett, M.H.; Hauser, A.R.; McColley, S.A.; Cullina, J.; Potter, E.; Powers, C.; Jain, M. Aminoglycoside resistance of Pseudomonas aeruginosa in cystic fibrosis results from convergent evolution in the mexZ gene. Thorax 2017, 72, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, Y.; Eda, S.; Gotoh, N.; Yoshihara, E.; Nakae, T. MexZ-mediated regulation of mexXY multidrug efflux pump expression in Pseudomonas aeruginosa by binding on the mexZ-mexX intergenic DNA. FEMS Microbiol. Lett. 2004, 238, 23–28. [Google Scholar] [PubMed]
- Morita, Y.; Sobel, M.L.; Poole, K. Antibiotic inducibility of the MexXY multidrug efflux system of Pseudomonas aeruginosa: Involvement of the antibiotic-inducible PA5471 gene product. J. Bacteriol. 2006, 188, 1847–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawalek, A.; Modrzejewska, M.; Zieniuk, B.; Bartosik, A.A.; Jagura-Burdzy, G. Interaction of ArmZ with the DNA-binding domain of MexZ induces expression of mexXY multidrug efflux pump genes and antimicrobial resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e01199-19. [Google Scholar] [CrossRef] [PubMed]
- Mena, A.; Smith, E.; Burns, J.; Speert, D.; Moskowitz, S.; Perez, J.; Oliver, A. Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J. Bacteriol. 2008, 190, 7910–7917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.E.; Buckley, D.G.; Wu, Z.; Saenphimmachak, C.; Hoffman, L.R.; D’Argenio, D.A.; Miller, S.I.; Ramsey, B.W.; Speert, D.P.; Moskowitz, S.M. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2006, 103, 8487–8492. [Google Scholar] [CrossRef] [Green Version]
- Llanes, C.; Hocquet, D.; Vogne, C.; Benali-Baitich, D.; Neuwirth, C.; Plésiat, P. Clinical strains of Pseudomonas aeruginosa overproducing MexAB-OprM and MexXY efflux pumps simultaneously. Antimicrob. Agents Chemother. 2004, 48, 1797–1802. [Google Scholar] [CrossRef] [Green Version]
- Guénard, S.; Muller, C.; Monlezun, L.; Benas, P.; Broutin, I.; Jeannot, K.; Plésiat, P. Multiple mutations lead to MexXY-OprM-dependent aminoglycoside resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Solé, M.; Fàbrega, A.; Cobos-Trigueros, N.; Zamorano, L.; Ferrer-Navarro, M.; Ballesté-Delpierre, C.; Reustle, A.; Castro, P.; Nicolás, J.M.; Oliver, A. In vivo evolution of resistance of Pseudomonas aeruginosa strains isolated from patients admitted to an intensive care unit: Mechanisms of resistance and antimicrobial exposure. J. Antimicrob. Chemother. 2015, 70, 3004–3013. [Google Scholar] [CrossRef] [Green Version]
- Vogne, C.; Aires, J.R.; Bailly, C.; Hocquet, D.; Plésiat, P. Role of the multidrug efflux system MexXY in the emergence of moderate resistance to aminoglycosides among Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob. Agents Chemother. 2004, 48, 1676–1680. [Google Scholar] [CrossRef] [Green Version]
- Masuda, N.; Sakagawa, E.; Ohya, S.; Gotoh, N.; Tsujimoto, H.; Nishino, T. Contribution of the MexX-MexY-OprM efflux system to intrinsic resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 2242–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westbrock-Wadman, S.; Sherman, D.R.; Hickey, M.J.; Coulter, S.N.; Zhu, Y.Q.; Warrener, P.; Nguyen, L.Y.; Shawar, R.M.; Folger, K.R.; Stover, C.K. Characterization of a Pseudomonas aeruginosa efflux pump contributing to aminoglycoside impermeability. Antimicrob. Agents Chemother. 1999, 43, 2975–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maunders, E.A.; Triniman, R.C.; Western, J.; Rahman, T.; Welch, M. Global reprogramming of virulence and antibiotic resistance in Pseudomonas aeruginosa by a single nucleotide polymorphism in elongation factor, fusA1. J. Biol. Chem. 2020, 295, 16411–16426. [Google Scholar] [CrossRef] [PubMed]
- Bolard, A.; Plésiat, P.; Jeannot, K. Mutations in gene fusA1 as a novel mechanism of aminoglycoside resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 62, e01835-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Causapé, C.; Sommer, L.M.; Cabot, G.; Rubio, R.; Ocampo-Sosa, A.A.; Johansen, H.K.; Figuerola, J.; Cantón, R.; Kidd, T.J.; Molin, S. Evolution of the Pseudomonas aeruginosa mutational resistome in an international cystic fibrosis clone. Sci. Rep. 2017, 7, 5555. [Google Scholar] [CrossRef] [Green Version]
- Farshadzadeh, Z.; Khosravi, A.D.; Alavi, S.M.; Parhizgari, N.; Hoveizavi, H. Spread of extended-spectrum β-lactamase genes of bla OXA-10, bla PER-1 and bla CTX-M in Pseudomonas aeruginosa strains isolated from burn patients. Burns 2014, 40, 1575–1580. [Google Scholar] [CrossRef]
- Jabalameli, F.; Taki, E.; Emaneini, M.; Beigverdi, R. Prevalence of metallo-β-lactamase-encoding genes among carbapenem-resistant Pseudomonas aeruginosa strains isolated from burn patients in Iran. Rev. Soc. Bras. Med. Trop. 2018, 51, 270–276. [Google Scholar] [CrossRef]
- Ahmadian, L.; Norouzi Bazgir, Z.; Ahanjan, M.; Valadan, R.; Goli, H.R. Role of Aminoglycoside-Modifying Enzymes (AMEs) in Resistance to Aminoglycosides among Clinical Isolates of Pseudomonas aeruginosa in the North of Iran. BioMed Res. Int. 2021, 2021, 7077344. [Google Scholar] [CrossRef]
- Poole, K. Pseudomonas aeruginosa: Resistance to the max. Front. Microbiol. 2011, 2, 65. [Google Scholar] [CrossRef] [Green Version]
- Kung, V.L.; Ozer, E.A.; Hauser, A.R. The accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2010, 74, 621–641. [Google Scholar] [CrossRef] [Green Version]
- Botelho, J.; Grosso, F.; Peixe, L. Antibiotic resistance in Pseudomonas aeruginosa—Mechanisms, epidemiology and evolution. Drug Resist. Updates 2019, 44, 100640. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.; Amyes, S. Oxa ß-lactamase. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freschi, L.; Bertelli, C.; Jeukens, J.; Moore, M.P.; Kukavica-Ibrulj, I.; Emond-Rheault, J.-G.; Hamel, J.; Fothergill, J.L.; Tucker, N.P.; McClean, S. Genomic characterisation of an international Pseudomonas aeruginosa reference panel indicates that the two major groups draw upon distinct mobile gene pools. FEMS Microbiol. Lett. 2018, 365, fny120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freschi, L.; Vincent, A.T.; Jeukens, J.; Emond-Rheault, J.-G.; Kukavica-Ibrulj, I.; Dupont, M.-J.; Charette, S.J.; Boyle, B.; Levesque, R.C. The Pseudomonas aeruginosa pan-genome provides new insights on its population structure, horizontal gene transfer, and pathogenicity. Genome Biol. Evol. 2019, 11, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.G.; Urbach, J.M.; Wu, G.; Liberati, N.T.; Feinbaum, R.L.; Miyata, S.; Diggins, L.T.; He, J.; Saucier, M.; Déziel, E. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 2006, 7, R90. [Google Scholar] [CrossRef] [Green Version]
- Bodendoerfer, E.; Marchesi, M.; Imkamp, F.; Courvalin, P.; Böttger, E.C.; Mancini, S. Co-occurrence of aminoglycoside and β-lactam resistance mechanisms in aminoglycoside-non-susceptible Escherichia coli isolated in the Zurich area, Switzerland. Int. J. Antimicrob. Agents 2020, 56, 106019. [Google Scholar] [CrossRef]
- Cox, G.; Stogios, P.J.; Savchenko, A.; Wright, G.D. Structural and molecular basis for resistance to aminoglycoside antibiotics by the adenylyltransferase ANT (2″)-Ia. MBio 2015, 6, e02180-14. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.S.; Nikolaidis, N.; Tolmasky, M.E. Rise and dissemination of aminoglycoside resistance: The aac (6′)-Ib paradigm. Front. Microbiol. 2013, 4, 121. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, F.; Peerayeh, S.N.; Nejad, Q.B.; Farhadian, A. The prevalence of aminoglycoside-modifying enzyme genes (aac (6′)-I, aac (6′)-II, ant (2”)-I, aph (3′)-VI) in Pseudomonas aeruginosa. Clinics 2011, 66, 1519–1522. [Google Scholar]
- Mendes, R.E.; Castanheira, M.; Toleman, M.A.; Sader, H.S.; Jones, R.N.; Walsh, T.R. Characterization of an integron carrying bla IMP-1 and a new aminoglycoside resistance gene, aac (6′)-31, and its dissemination among genetically unrelated clinical isolates in a Brazilian hospital. Antimicrob. Agents Chemother. 2007, 51, 2611–2614. [Google Scholar] [CrossRef] [Green Version]
- Sobel, M.L.; McKay, G.A.; Poole, K. Contribution of the MexXY multidrug transporter to aminoglycoside resistance in Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 2003, 47, 3202–3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thacharodi, A.; Lamont, I.L. Aminoglycoside resistance in Pseudomonas aeruginosa: The contribution of the MexXY-OprM efflux pump varies between isolates. J. Med. Microbiol. 2022; in press. [Google Scholar]
- Qiu, X.; Kulasekara, B.; Lory, S. Role of horizontal gene transfer in the evolution of Pseudomonas aeruginosa virulence. In Microbial Pathogenomics; Karger Publishers: Basel, Switzerland, 2009; Volume 6, pp. 126–139. [Google Scholar]
- Ramírez, M.S.; Quiroga, C.; Centrón, D. Novel rearrangement of a class 2 integron in two non-epidemiologically related isolates of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2005, 49, 5179–5181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakulenko, S.B.; Mobashery, S. Versatility of aminoglycosides and prospects for their future. Clin. Microbiol. Rev. 2003, 16, 430–450. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updates 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettner, M.; Kallova, J.; Hletkova, M.; Milošovič, P. Incidence and mechanisms of aminoglycoside resistance in Pseudomonas aeruginosa serotype O11 isolates. Infection 1995, 23, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.; Perlin, M.H.; Baquero, F.; Lerner, D.L.; Lerner, S.A. High-level amikacin resistance in Pseudomonas aeruginosa associated with a 3′-phosphotransferase with high affinity for amikacin. Int. J. Antimicrob. Agents 2000, 15, 257–263. [Google Scholar] [CrossRef]
- Lambert, T.; Gerbaud, G.; Courvalin, P. Characterization of transposon Tn1528, which confers amikacin resistance by synthesis of aminoglycoside 3′-O-phosphotransferase type VI. Antimicrob. Agents Chemother. 1994, 38, 702–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atassi, G.; Scheetz, M.H.; Nozick, S.; Rhodes, N.J.; Murphy-Belcaster, M.; Murphy, K.R.; Ozer, E.A.; Hauser, A.R. Genomics of Aminoglycoside Resistance in Pseudomonas aeruginosa Bloodstream Infections at a United States Academic Hospital. Medrxiv 2021. [Google Scholar] [CrossRef]
- Seupt, A.; Schniederjans, M.; Tomasch, J.; Häussler, S. Expression of the MexXY aminoglycoside efflux pump and presence of an aminoglycoside-modifying enzyme in clinical Pseudomonas aeruginosa isolates are highly correlated. Antimicrob. Agents Chemother. 2020, 65, e01166-20. [Google Scholar] [CrossRef]
- Hirsch, D.R.; Cox, G.; D’Erasmo, M.P.; Shakya, T.; Meck, C.; Mohd, N.; Wright, G.D.; Murelli, R.P. Inhibition of the ANT (2″)-Ia resistance enzyme and rescue of aminoglycoside antibiotic activity by synthetic α-hydroxytropolones. Bioorg. Med. Chem. Lett. 2014, 24, 4943–4947. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.H. Experiments in Molecular Genetics; Cold Spring Harbor Laboratory: New York, NY, USA, 1972; pp. 352–355. [Google Scholar]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- CLSI. Performance standards for antimicrobial susceptibility testing, 28th ed.; Clinical and Laboratory Standards Insitute: Wayne, PA, USA, 2018. [Google Scholar]
- Sambrook, J.; Russel, D. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; Volume 1, pp. 5.26–5.28. [Google Scholar]
- Hmelo, L.R.; Borlee, B.R.; Almblad, H.; Love, M.E.; Randall, T.E.; Tseng, B.S.; Lin, C.; Irie, Y.; Storek, K.M.; Yang, J.J. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat. Protoc. 2015, 10, 1820–1841. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.T.; Karkhoff-Schweizer, R.R.; Kutchma, A.J.; Schweizer, H.P. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: Application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 1998, 212, 77–86. [Google Scholar] [CrossRef]
- Thoma, S.; Schobert, M. An improved Escherichia coli donor strain for diparental mating. FEMS Microbiol. Lett. 2009, 294, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Baynham, P.J.; Ramsey, D.M.; Gvozdyev, B.V.; Cordonnier, E.M.; Wozniak, D.J. The Pseudomonas aeruginosa ribbon-helix-helix DNA-binding protein AlgZ (AmrZ) controls twitching motility and biogenesis of type IV pili. J. Bacteriol. 2006, 188, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Hilliam, Y.; Moore, M.P.; Lamont, I.L.; Bilton, D.; Haworth, C.S.; Foweraker, J.; Walshaw, M.J.; Williams, D.; Fothergill, J.L.; De Soyza, A. Pseudomonas aeruginosa adaptation and diversification in the non-cystic fibrosis bronchiectasis lung. Eur. Respir. J. 2017, 49, 1602108. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.W.; Robson, C.L.; Watts, A.M.; Gray, A.R.; Wainwright, C.E.; Bell, S.C.; Ramsay, K.A.; Kidd, T.J.; Reid, D.W.; Brockway, B. Expression of Pseudomonas aeruginosa antibiotic resistance genes varies greatly during infections in cystic fibrosis patients. Antimicrob. Agents Chemother. 2018, 62, e01789-18. [Google Scholar] [CrossRef] [Green Version]
- Wardell, S.J.; Rehman, A.; Martin, L.W.; Winstanley, C.; Patrick, W.M.; Lamont, I.L. A large-scale whole-genome comparison shows that experimental evolution in response to antibiotics predicts changes in naturally evolved clinical Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e01619-19. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Subedi, D.; Vijay, A.K.; Kohli, G.S.; Rice, S.A.; Willcox, M. Comparative genomics of clinical strains of Pseudomonas aeruginosa strains isolated from different geographic sites. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.H.; Tetu, S.G.; Larouche, A.; Elbourne, L.; Tremblay, S.; Ren, Q.; Dodson, R.; Harkins, D.; Shay, R.; Watkins, K. Complete genome sequence of the multiresistant taxonomic outlier Pseudomonas aeruginosa PA7. PLoS ONE 2010, 5, e8842. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; Garcìa-Fernandez, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. PlasmidFinder and pMLST: In silico detection and typing of plasmids. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.H.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 2021, 76, 101–109. [Google Scholar] [CrossRef]
- Yoshii, A.; Moriyama, H.; Fukuhara, T. The novel kasugamycin 2′-N-acetyltransferase gene aac (2′)-IIa, carried by the IncP island, confers kasugamycin resistance to rice-pathogenic bacteria. Appl. Environ. Microbiol. 2012, 78, 5555–5564. [Google Scholar] [CrossRef] [Green Version]
- Haines, A.S.; Jones, K.; Cheung, M.; Thomas, C.M. The IncP-6 plasmid Rms149 consists of a small mobilizable backbone with multiple large insertions. J. Bacteriol. 2005, 187, 4728–4738. [Google Scholar] [CrossRef] [Green Version]
- Gibb, A.P.; Tribuddharat, C.; Moore, R.A.; Louie, T.J.; Krulicki, W.; Livermore, D.M.; Palepou, M.-F.I.; Woodford, N. Nosocomial outbreak of carbapenem-resistant Pseudomonas aeruginosa with a new bla IMP allele, bla IMP-7. Antimicrob. Agents Chemother. 2002, 46, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Tenover, F.C.; Phillips, K.; Gilbert, T.; Lockhart, P.; O’Hara, P.; Plorde, J. Development of a DNA probe from the deoxyribonucleotide sequence of a 3-N-aminoglycoside acetyltransferase [AAC (3)-I] resistance gene. Antimicrob. Agents Chemother. 1989, 33, 551–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, D.L.; Nelson, L.E.; Shawar, R.M.; Lin, B.B.; Lockwood, L.G.; Dirks, J.E.; Miller, G.H.; Burns, J.L.; Garber, R.L. Aminoglycoside-resistance mechanisms for cystic fibrosis Pseudomonas aeruginosa isolates are unchanged by long-term, intermittent, inhaled tobramycin treatment. J. Infect. Dis. 2000, 181, 1180–1184. [Google Scholar] [CrossRef] [Green Version]
- Vliegenthart, J.; Ketelaar-van Gaalen, P.; van de Klundert, J. Nucleotide sequence of the aacC3 gene, a gentamicin resistance determinant encoding aminoglycoside-(3)-N-acetyltransferase III expressed in Pseudomonas aeruginosa but not in Escherichia coli. Antimicrob. Agents Chemother. 1991, 35, 892–897. [Google Scholar] [CrossRef] [Green Version]
- Plattner, M.; Gysin, M.; Haldimann, K.; Becker, K.; Hobbie, S.N. Epidemiologic, phenotypic, and structural characterization of aminoglycoside-resistance gene aac (3)-IV. Int. J. Mol. Sci. 2020, 21, 6133. [Google Scholar] [CrossRef] [PubMed]
- Casin, I.; Bordon, F.; Bertin, P.; Coutrot, A.; Podglajen, I.; Brasseur, R.; Collatz, E. Aminoglycoside 6′-N-acetyltransferase variants of the Ib type with altered substrate profile in clinical isolates of Enterobacter cloacae and Citrobacter freundii. Antimicrob. Agents Chemother. 1998, 42, 209–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rather, P.; Munayyer, H.; Mann, P.; Hare, R.; Miller, G.; Shaw, K. Genetic analysis of bacterial acetyltransferases: Identification of amino acids determining the specificities of the aminoglycoside 6’-N-acetyltransferase Ib and IIa proteins. J. Bacteriol. 1992, 174, 3196–3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viedma, E.; Juan, C.; Acosta, J.; Zamorano, L.; Otero, J.R.; Sanz, F.; Chaves, F.; Oliver, A. Nosocomial spread of colistin-only-sensitive sequence type 235 Pseudomonas aeruginosa isolates producing the extended-spectrum β-lactamases GES-1 and GES-5 in Spain. Antimicrob. Agents Chemother. 2009, 53, 4930–4933. [Google Scholar] [CrossRef] [Green Version]
- Hollingshead, S.; Vapnek, D. Nucleotide sequence analysis of a gene encoding a streptomycin/spectinomycin adenyltransferase. Plasmid 1985, 13, 17–30. [Google Scholar] [CrossRef]
- Gu, B.; Tong, M.; Zhao, W.; Liu, G.; Ning, M.; Pan, S.; Zhao, W. Prevalence and characterization of class I integrons among Pseudomonas aeruginosa and Acinetobacter baumannii isolates from patients in Nanjing, China. J. Clin. Microbiol. 2007, 45, 241–243. [Google Scholar] [CrossRef] [Green Version]
- Kazama, H.; Kizu, K.; Iwasaki, M.; Hamashima, H.; Sasatsu, M.; Arai, T. A new gene, aadA2b, encoding an aminoglycoside adenylyltransferase, AAD (3″)(9), isolated from integron InC in Pseudomonas aeruginosa. Microbios 1996, 86, 77–83. [Google Scholar]
- Adrian, P.V.; Thomson, C.J.; Klugman, K.P.; Amyes, S.G. New gene cassettes for trimethoprim resistance, dfr13, and streptomycin-spectinomycin resistance, aadA4, inserted on a class 1 integron. Antimicrob. Agents Chemother. 2000, 44, 355–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadovasilaki, M.; Oberthür, D.; Gessmann, R.; Sarrou, I.; Betzel, C.; Scoulica, E.; Petratos, K. Biophysical and enzymatic properties of aminoglycoside adenylyltransferase AadA6 from Pseudomonas aeruginosa. Biochem. Biophys Rep. 2015, 4, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Santanam, P.; Kayser, F.H. Purification and characterization of an aminoglycoside inactivating enzyme from Staphylococcus epidermidis FK109 that nucleotidylates the 4’-and 4″-hydroxyl groups of the aminoglycoside antibiotics. J. Antibiot. 1978, 31, 343–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esparragón, F.R.; Martín, M.G.; Lama, Z.G.; Sabatelli, F.J.; Junco, M.T.T. Aminoglycoside resistance mechanisms in clinical isolates of Pseudomonas aeruginosa from the Canary Islands. Zentralbl. Bakteriol. 2000, 289, 817–826. [Google Scholar] [CrossRef]
- Woegerbauer, M.; Kuffner, M.; Domingues, S.; Nielsen, K.M. Involvement of aph (3′)-IIa in the formation of mosaic aminoglycoside resistance genes in natural environments. Front. Microbiol. 2015, 6, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stogios, P.J.; Shakya, T.; Evdokimova, E.; Savchenko, A.; Wright, G.D. Structure and function of APH (4)-Ia, a hygromycin B resistance enzyme. J. Biol. Chem. 2011, 286, 1966–1975. [Google Scholar] [CrossRef] [Green Version]
- Ashenafi, M.; Ammosova, T.; Nekhai, S.; Byrnes, W.M. Purification and characterization of aminoglycoside phosphotransferase APH (6)-Id, a streptomycin-inactivating enzyme. Mol. Cell Biochem. 2014, 387, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Stover, C.K.; Pham, X.Q.; Erwin, A.; Mizoguchi, S.; Warrener, P.; Hickey, M.; Brinkman, F.; Hufnagle, W.; Kowalik, D.; Lagrou, M. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef]
- Rehman, A.; Jeukens, J.; Levesque, R.C.; Lamont, I.L. Gene-gene interactions dictate ciprofloxacin resistance in Pseudomonas aeruginosa and facilitate prediction of resistance phenotype from genome sequence data. Antimicrob. Agents Chemother. 2021, 65, e02696-20. [Google Scholar] [CrossRef]
- Yanisch-Perron, C.; Vieira, J.; Messing, J. Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mpl8 and pUC19 vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef]
- Freschi, L.; Jeukens, J.; Kukavica-Ibrulj, I.; Boyle, B.; Dupont, M.J.; Laroche, J.; Larose, S.; Maaroufi, H.; Fothergill, J.L.; Moore, M.; et al. Clinical utilization of genomics data produced by the international Pseudomonas aeruginosa consortium. Front. Microbiol. 2015, 6, 1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Isolates | Source | Sequence Type b | MIC a | Sequence Variants | Other Acquired AMEs | ||
|---|---|---|---|---|---|---|---|
| Tob | Gen | Amik | |||||
| Isolates with Ant (2″)-Ia | |||||||
| 403-105 | CF | 775 | 1024 | 512 | 16 | mexZ (A38T) armZ (H182Q) fusA1 (Y690C) | |
| 008-A1 | CF | 775 | 512 | 128 | 8 | mexZ (A38T) armZ (H182Q) fusA1 (Y690C) | |
| 1257147 | Bladder | 235 | 512 | 256 | 512 | armZ (H182Q) | Aac (6′)-Ib Aph (6)-Id Aph (3″)-Ib |
| 1268230 | Wound | 175 | 32 | 16 | 4 | armZ (H182Q) mexZ (G195E) | |
| 1271701 | Urine | 1560 | 16 | 32 | 8 | armZ (H182Q) | Aph (3′)-IIb AadA1 (ANT (3″)) |
| 1324459 | Burn | 357 | 128 | 128 | 128 | Aac (6′)-11 AadA1(ANT (3″)) | |
| 1607533 | Colon | 234 | 2 | 2 | 4 | mexY (E592K) | Aph (3′)- IIb Aph (3″)-Ib Aph (6)-Id |
| Isolates with Aac (6′)-Ib3 | |||||||
| 1260990 | Urine | 395 | 32 | 64 | 128 | armZ (H182Q) | AadA6 (ANT (3″)) Aph (3′) VIa |
| 1275655 | Wound | 235 | 16 | 2 | 16 | armZ(H182Q) mexXY(Stop codon) | Aac (6′)-33 AadA1b Aph (3′)-IIa Aph (6)-Ic |
| 1295835 | Sputum | 646 | 128 | 128 | 32 | armZ (H182Q) | |
| 1344658 | Respiratory: Endotracheal aspirate | 292 | 256 | 512 | 256 | armZ (H182Q) amgS (P139S) | AadA2b Aph (3″)-Ib Aph (6)-Id |
| 1420275 | Respiratory: Endotracheal aspirate | 309 | 256 | 256 | 256 | mexZ (∆6 bp) | Aac (6′)-33 AadA1b |
| 1586994 | Blood | 235 | 256 | 128 | 256 | armZ (H182Q) mexZ (∆93 bp) | AadA6 Aac (6′)-Ib-cr |
| 1690076 | Respiratory: Endotracheal aspirate | 309 | 256 | 128 | 256 | mexZ (∆6 bp) | Aac (6′)-33 AadA1b |
| Isolate with Aph (3′)-VIa | |||||||
| 1260990 | Urine | 395 | 32 | 64 | 128 | armZ (H182Q) | AadA6 (ANT (3″)) Aac (6′)-Ib3 |
| Wildtype Isolate | ∆mexXY Mutant | ∆mexXY Mutant [ant (2″)-Ia] | Fold A | ∆mexXY Mutant [aac (6′)-Ib3] | Fold | ∆mexXY Mutant [aph (3′)-VIa] | Fold | |
|---|---|---|---|---|---|---|---|---|
| PAO1 | ||||||||
| Tob B | 0.5 C | 0.25 | 8 | 32 | 16 | 64 | 0.25 | 0 |
| Gen | 1 | 0.25 | 16 | 64 | 4 | 16 | 0.25 | 0 |
| Amik | 2 | 0.5 | 0.5 | 0 | 0.5 | 0 | 64 | 128 |
| 006A2 D | ||||||||
| Tob | 32 | 1 | 16 | 16 | 16 | 16 | 1 | 0 |
| Gen | 32 | 1 | 16 | 16 | 8 | 8 | 1 | 0 |
| Amik | 128 | 4 | 4 | 0 | 4 | 0 | 128 | 32 |
| 403-107 | ||||||||
| Tob | 2 | 0.5 | 8 | 16 | 8 | 16 | 0.5 | 0 |
| Gen | 4 | 0.25 | 8 | 32 | 4 | 16 | 0.25 | 0 |
| Amik | 16 | 1 | 1 | 0 | 1 | 0 | 32 | 32 |
| 015A | ||||||||
| Tob | 8 | 2 | 8 | 4 | 8 | 4 | 2 | 0 |
| Gen | 16 | 1 | 8 | 8 | 4 | 4 | 1 | 0 |
| Amik | 32 | 4 | 4 | 0 | 4 | 0 | 64 | 16 |
| Fold range | 4–64 | 4–64 | 16–128 | |||||
| Antibiotics | PAO1 | ∆mexZ | fusA1 (R680C) |
|---|---|---|---|
| Empty vector | |||
| Tob | 0.5 | 1 | 2 |
| Gen | 1 | 2 | 4 |
| Amik | 2 | 4 | 8 |
| Expressing ant (2″)-IA | |||
| Tob | 16 | 16 | 32 |
| Gen | 64 | 256 | 128 |
| Expressing acc (6′)-Ib3 | |||
| Tob | 32 | 32 | 32 |
| Gen | 32 | 64 | 32 |
| Expressing aph (3′)-VIa | |||
| Amik | 128 | 512 | 256 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thacharodi, A.; Lamont, I.L. Aminoglycoside-Modifying Enzymes Are Sufficient to Make Pseudomonas aeruginosa Clinically Resistant to Key Antibiotics. Antibiotics 2022, 11, 884. https://doi.org/10.3390/antibiotics11070884
Thacharodi A, Lamont IL. Aminoglycoside-Modifying Enzymes Are Sufficient to Make Pseudomonas aeruginosa Clinically Resistant to Key Antibiotics. Antibiotics. 2022; 11(7):884. https://doi.org/10.3390/antibiotics11070884
Chicago/Turabian StyleThacharodi, Aswin, and Iain L. Lamont. 2022. "Aminoglycoside-Modifying Enzymes Are Sufficient to Make Pseudomonas aeruginosa Clinically Resistant to Key Antibiotics" Antibiotics 11, no. 7: 884. https://doi.org/10.3390/antibiotics11070884