
TXH11106: A Third-Generation MreB Inhibitor with Enhanced Activity against a Broad Range of Gram-Negative Bacterial Pathogens

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Other Reagents
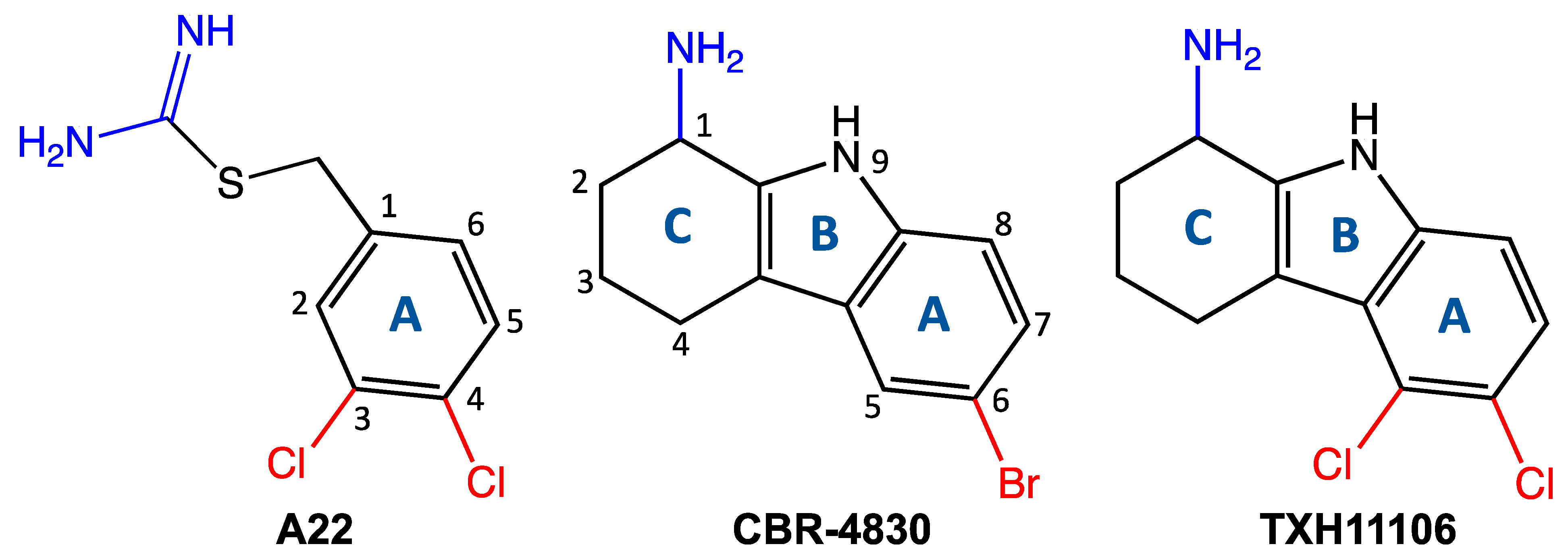
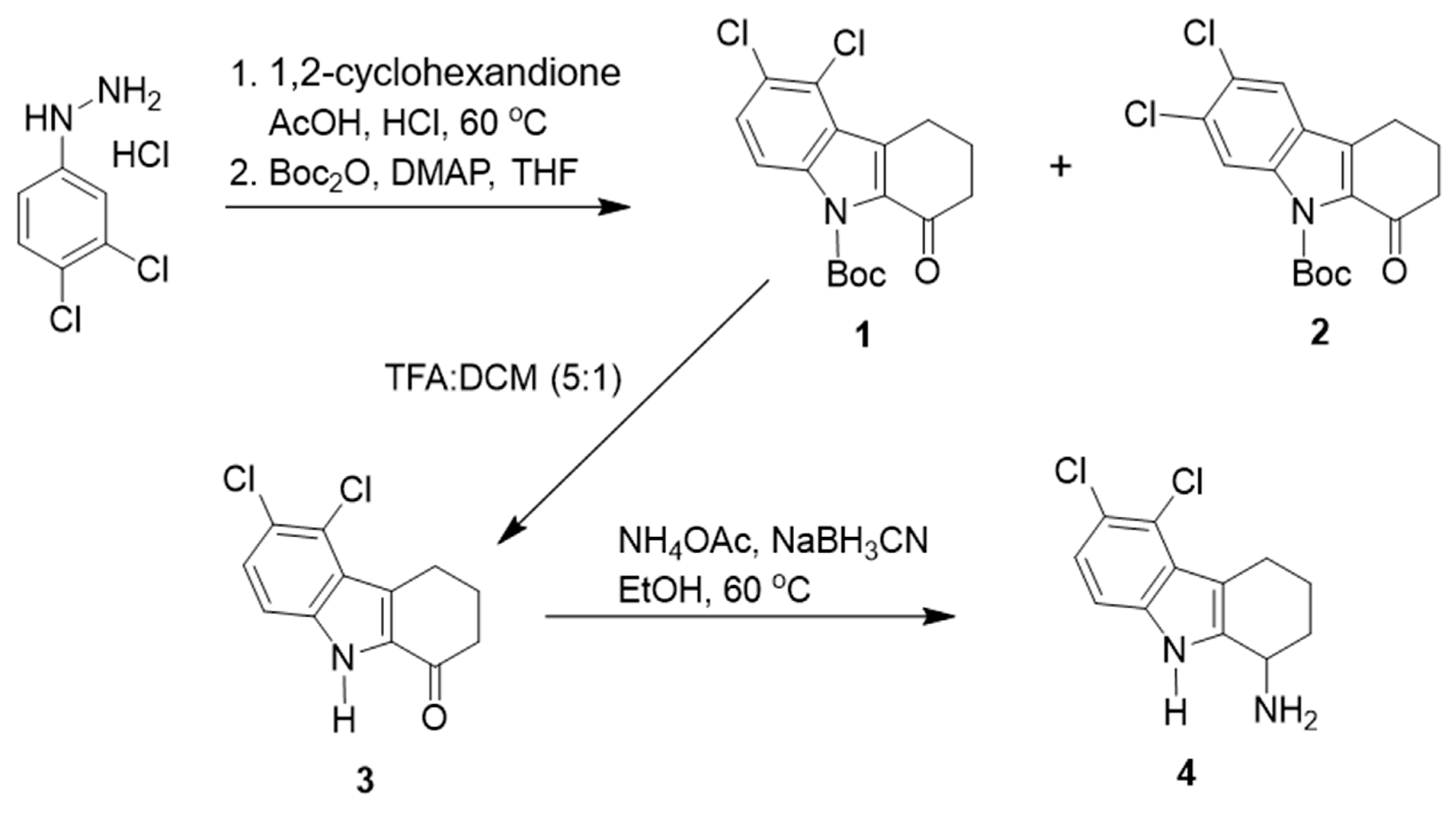
2.2. Synthesis of TXH11106 (4)
- (i)
- 5,6-Dichloro-2,3,4,9-tetrahydro-1H-carbazol-1-one (3):
- (ii)
- tert-Butyl 5,6-dichloro-1-oxo-1,2,3,4-tetrahydro-9H-carbazole-9-carboxylate (1):
2.3. MIC and MBC Assays
2.4. Time-Dependent Kill Assay
2.5. Assay for Frequency of Resistance (FOR)
2.6. Differential Interference Contrast (DIC) Microscopy
2.7. Cloning, Expression, and Purification of Wild-Type (WT) and E143G Mutant E. coli MreB
2.8. ATPase Assays
3. Results
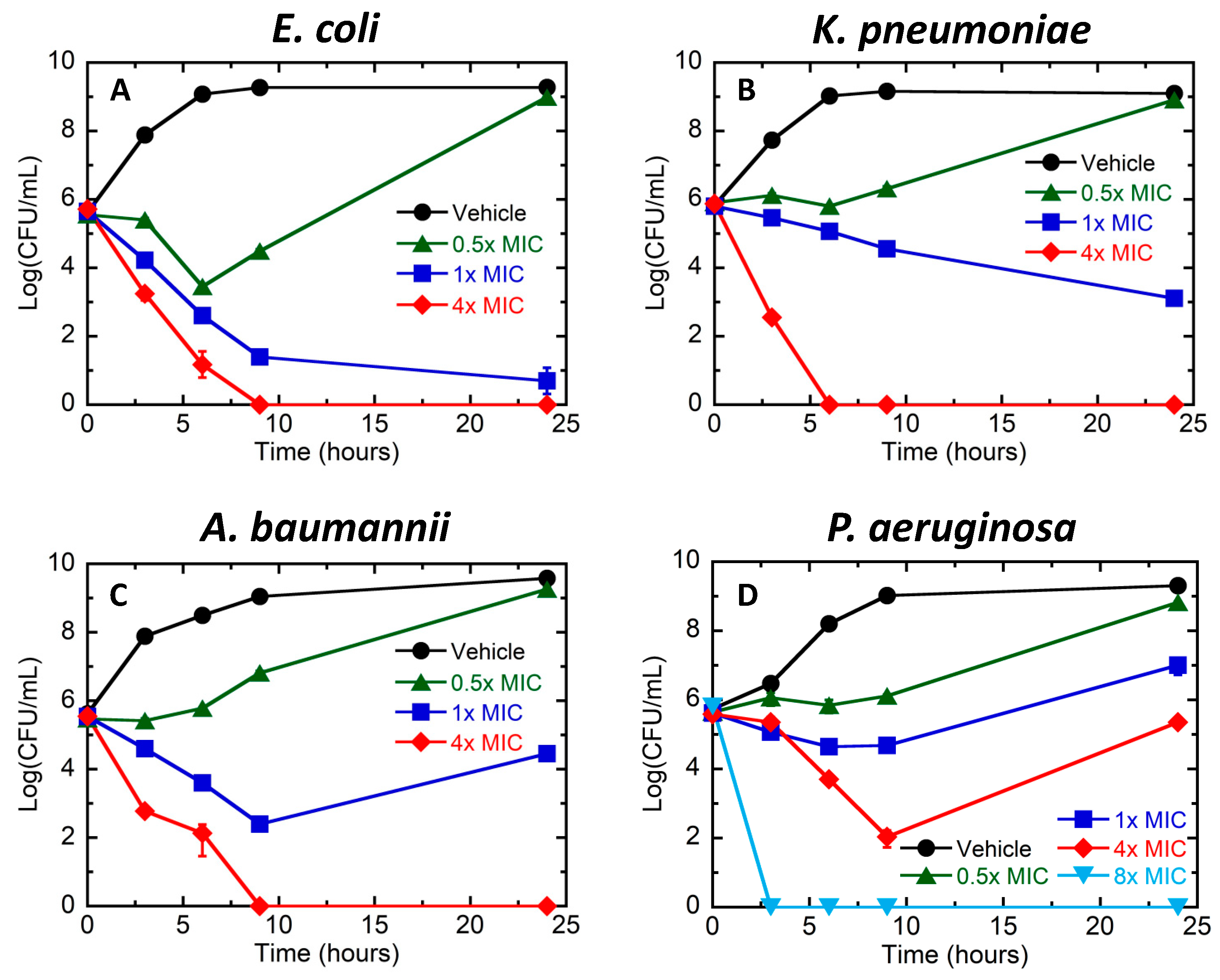
3.1. Bactericidal Activity of TXH11106 against the Gram-Negative Pathogens E. coli, K. pneumoniae, A. baumannii, and P. aeruginosa
3.2. Frequency of Resistance (FOR) in E. coli, K. pneumoniae, A. baumannii, and P. aeruginosa Following Exposure to TXH11106
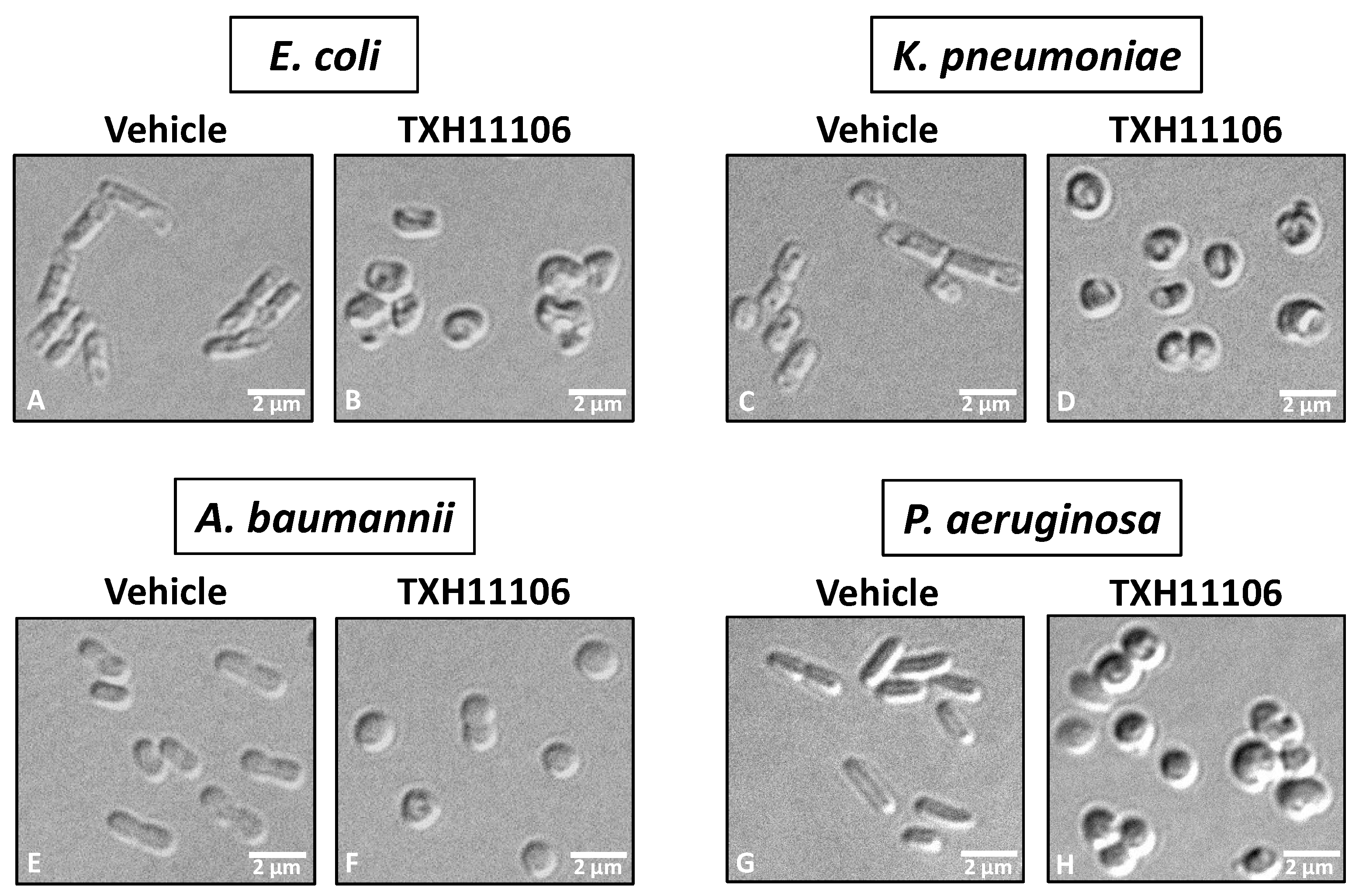
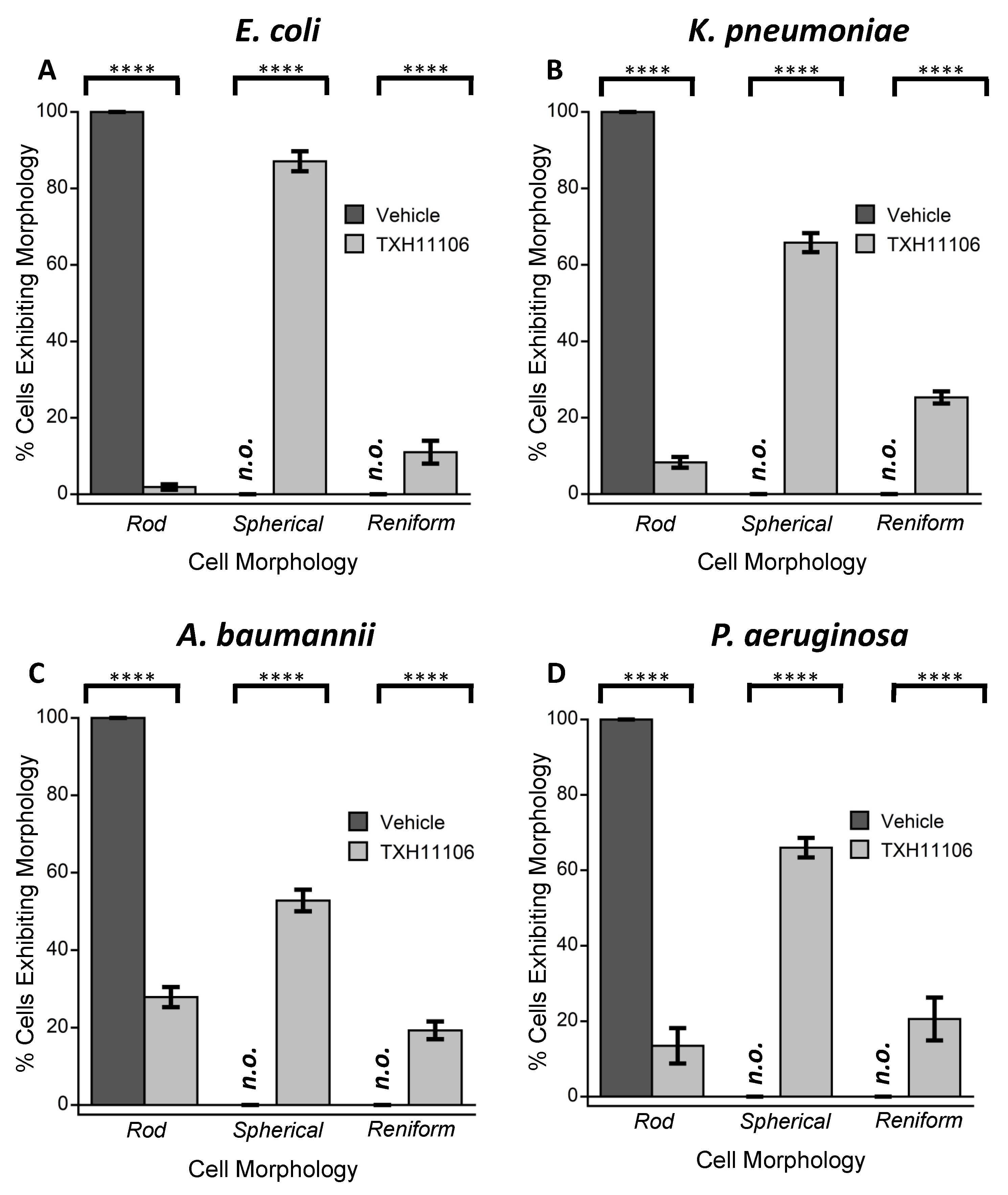
3.3. Impact of TXH11106 Treatment on the Morphology of E. coli, K. pneumoniae, A. baumannii, and P. aeruginosa
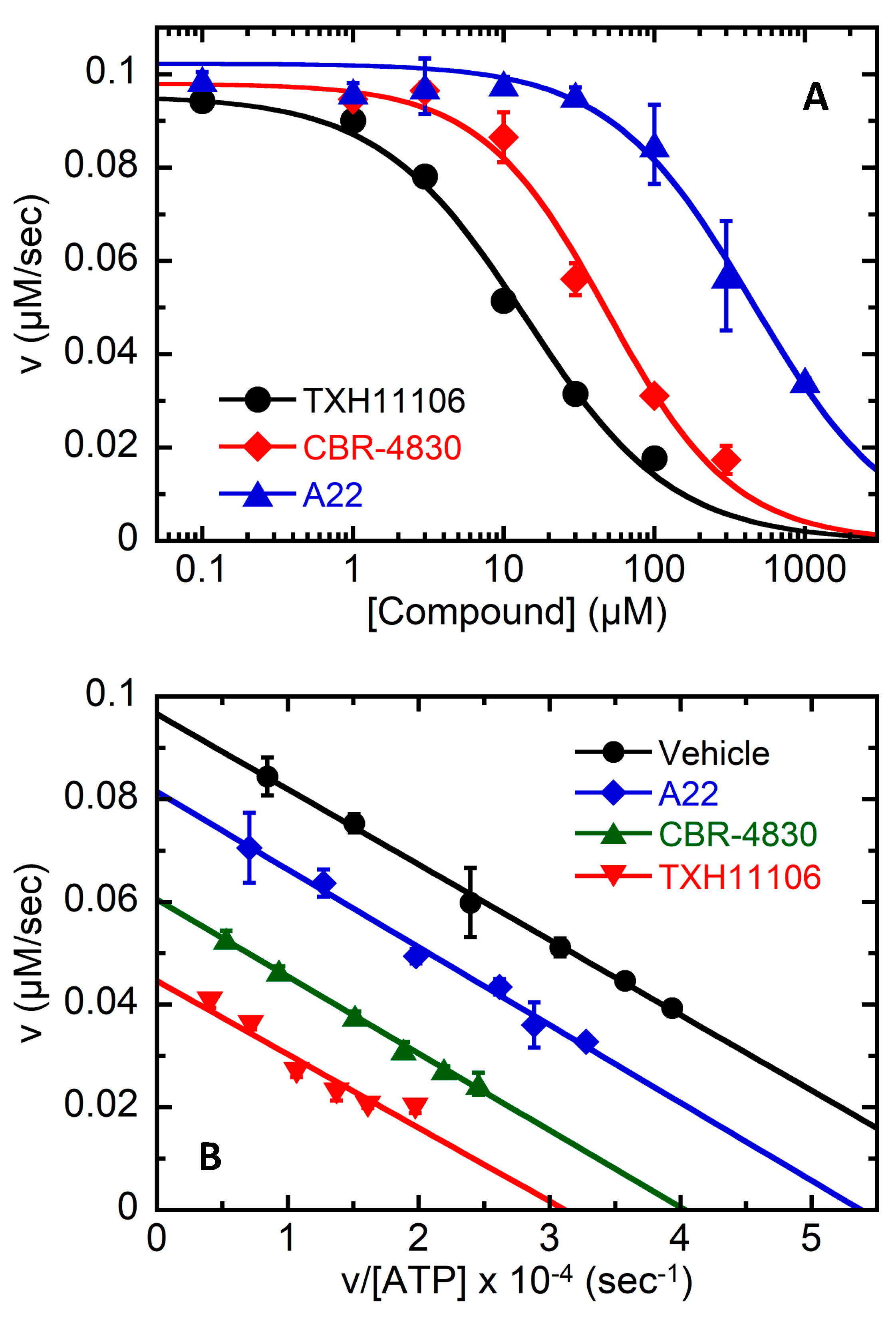
3.4. TXH11106-Induced Inhibition of the ATPase Activity of E. coli MreB (EcMreB)
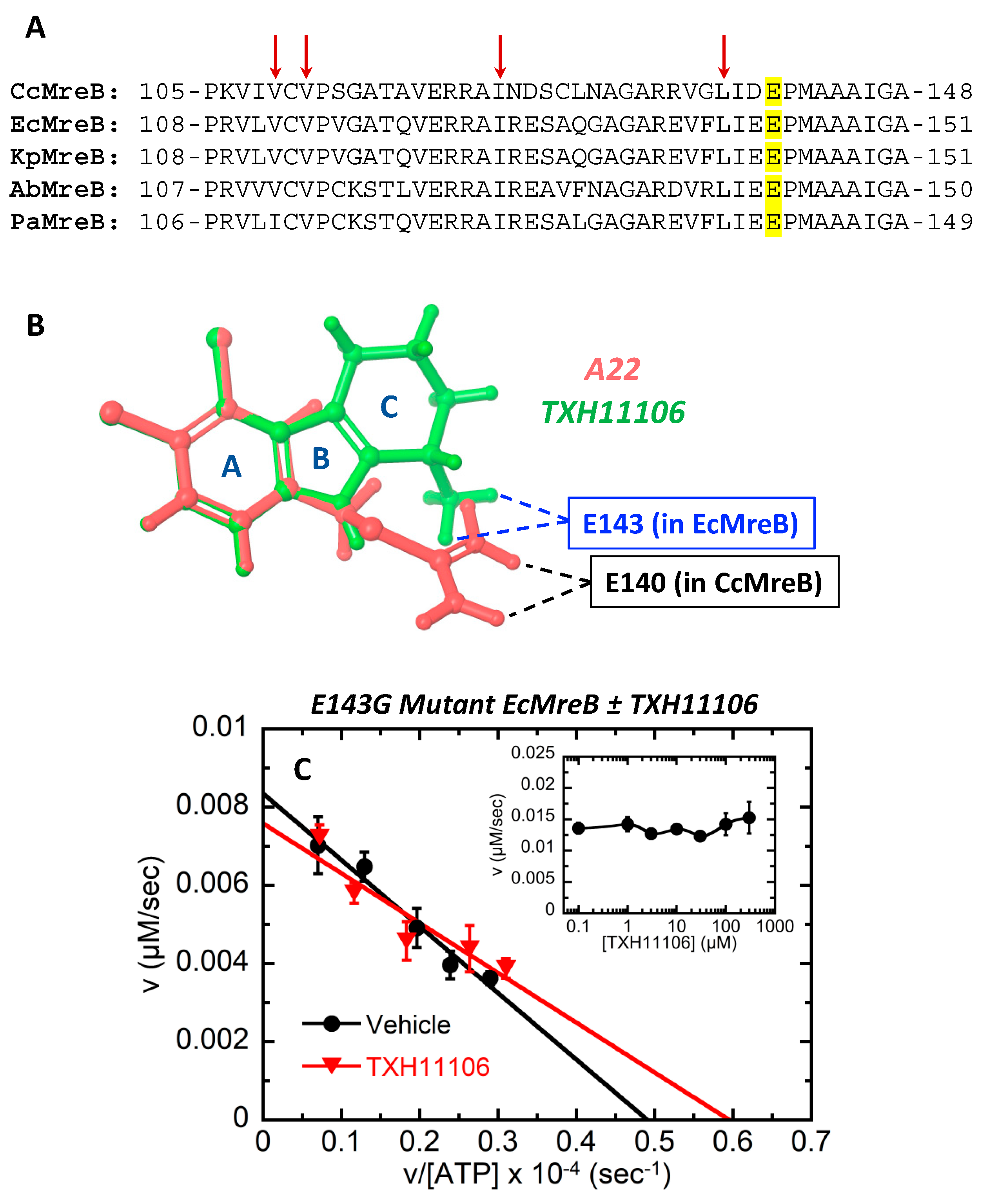
3.5. Impact of the E143G Mutation on the Catalytic Activity of EcMreB as Well as Inhibition by TXH11106
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nature, E. The Antibiotic Alarm. Nature 2013, 495, 141. [Google Scholar] [CrossRef]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Causes of Antibiotic Resistance; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Medina, E.; Pieper, D.H. Tackling Threats and Future Problems of Multidrug-Resistant Bacteria. In How to Overcome the Antibiotic Crisis: Facts, Challenges, Technologies and Future Perspectives; Stadler, M., Dersch, P., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 3–33. [Google Scholar]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2019; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2019.
- Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of gram-negative bacteria to current antibacterial agents and approaches to resolve it. Molecules 2020, 25, 1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakoullis, L.; Papachristodoulou, E.; Chra, P.; Panos, G. Mechanisms of antibiotic resistance in important gram-positive and gram-negative pathogens and novel antibiotic solutions. Antibiotics 2021, 10, 415. [Google Scholar] [CrossRef]
- Kapoor, G.; Saigal, S.; Elongavan, A. Action and resistance mechanisms of antibiotics: A guide for clinicians. J. Anaesthesiol. Clin. Pharmacol. 2017, 33, 300. [Google Scholar] [CrossRef]
- Awuni, E. Status of Targeting MreB for the Development of Antibiotics. Front. Chem. 2020, 7, 884. [Google Scholar] [CrossRef] [Green Version]
- Belete, T.M. Novel targets to develop new antibacterial agents and novel alternatives to antibacterial agents. Hum. Microbiome J. 2019, 11, 100052. [Google Scholar] [CrossRef]
- Han, H.; Wang, Z.; Li, T.; Teng, D.; Mao, R.; Hao, Y.; Yang, N.; Wang, X.; Wang, J. Recent progress of bacterial FtsZ inhibitors with a focus on peptides. FEBS J. 2021, 288, 1091–1106. [Google Scholar] [CrossRef]
- Luther, A.; Urfer, M.; Zahn, M.; Müller, M.; Wang, S.-Y.; Mondal, M.; Vitale, A.; Hartmann, J.-B.; Sharpe, T.; Monte, F.L.; et al. Chimeric peptidomimetic antibiotics against gram-negative bacteria. Nature 2019, 576, 452–458. [Google Scholar] [CrossRef]
- Figge, R.M.; Divakaruni, A.V.; Gober, J.W. MreB, the cell shape-determining bacterial actin homologue, co-ordinates cell wall morphogenesis in Caulobacter crescentus. Mol. Microbiol. 2004, 51, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- van den Ent, F.; Amos, L.A.; Löwe, J. Prokaryotic origin of the actin cytoskeleton. Nature 2001, 413, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H. The sugar kinase/heat shock protein 70/Actin superfamily: Implications of conserved structure for mechanism. Annu. Rev. Biophys. Biomol. Struct. 1996, 25, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Den Blaauwen, T.; De Pedro, M.A.; Nguyen-Distèche, M.; Ayala, J.A. Morphogenesis of rod-shaped sacculi. FEMS Microbiol. Rev. 2008, 32, 321–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Normark, S. Mutation in Escherichia coli K-12 Mediating Spherelike envelopes and changes tolerance to ultraviolet irradiation and some antibiotics. J. Bacteriol. 1969, 98, 1274–1277. [Google Scholar] [CrossRef] [Green Version]
- Wachi, M.; Doi, M.; Tamaki, S.; Park, W.; Nakajima-Iijima, S.; Matsuhashi, M. Mutant isolation and molecular cloning of mre Genes, which determine cell shape, sensitivity to Mecillinam, and amount of penicillin-binding proteins in Escherichia coli. J. Bacteriol. 1987, 169, 4935–4940. [Google Scholar] [CrossRef] [Green Version]
- van der Ploeg, R.; Verheul, J.; Vischer, N.O.E.; Alexeeva, S.; Hoogendoorn, E.; Postma, M.; Banzhaf, M.; Vollmer, W.; den Blaauwen, T. Colocalization and interaction between elongasome and divisome during a preparative cell division phase in Escherichia coli. Mol. Microbiol. 2013, 87, 1074–1087. [Google Scholar] [CrossRef]
- Iwai, N.; Nagai, K.; Wachi, M. Novel S-Benzylisothiourea compound that induces spherical cells in Escherichia coli probably by acting on a rod-shape-determining protein(s) other than penicillin-binding protein 2. Biosci. Biotechnol. Biochem. 2002, 66, 2658–2662. [Google Scholar] [CrossRef] [Green Version]
- Bonez, P.C.; Ramos, A.P.; Nascimento, K.; Copetti, P.M.; Souza, M.E.; Rossi, G.G.; Agertt, V.A.; Sagrillo, M.R.; Santos, R.C.V.; Campos, M.M.A. Antibacterial, Cyto and genotoxic activities of a22 compound ((S-3,4-dichlorobenzyl)isothiourea hydrochloride). Microb. Pathog. 2016, 99, 14–18. [Google Scholar] [CrossRef]
- van den Ent, F.; Izoré, T.; Bharat, T.A.; Johnson, C.M.; Löwe, J. Bacterial actin MreB forms antiparallel double filaments. eLife 2014, 3, e02634. [Google Scholar] [CrossRef]
- Robertson, G.T.; Doyle, T.B.; Du, Q.; Duncan, L.; Mdluli, K.E.; Lynch, A.S. A novel indole compound that inhibits Pseudomonas aeruginosa growth by targeting MreB is a Substrate for MexAB-OprM. J. Bacteriol. 2007, 189, 6870–6881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 11th ed.; CLSI Standard M07: Wayne, PA, USA, 2018. [Google Scholar]
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. Pharmacokinetics and in vivo Antistaphylococcal Efficacy of TXY541, a 1-methylpiperidine-4-carboxamide Prodrug of PC190723. Biochem. Pharmacol. 2013, 86, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Shigemura, K.; Arakawa, S.; Tanaka, K.; Fujisawa, M. Clinical investigation of isolated bacteria from urinary tracts of hospitalized patients and their susceptibilities to antibiotics. J. Infect. Chemother. 2009, 15, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Miles Hacker, K.B.; William, M. Pharmacology: Principles and Practice; Hacker, M., Ed.; Academic Press: Burlington, MA, USA, 2009; p. 608. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Antibacterial Parameters (MIC and MBC Listed in µg/mL) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E. coli 25922 | K. pneumoniae 13883 | A. baumannii 19606 | P. aeruginosa 27853 | |||||||||
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |||||
| TXH11106 | 4 | 4 | 1 | 8 | 8 | 1 | 8 | 16 | 2 | 8 | 32 | 4 |
| CBR-4830 | 8 | 8 | 1 | 16 | 64 | 4 | 32 | 64 | 2 | 32 | 128 | 4 |
| A22 | 256 | 256 | 1 | 128 | 256 | 2 | 32 | 128 | 4 | 32 | >256 | >8 |
| Strain | [TXH11106] (µg/mL) | FOR |
|---|---|---|
| E. coli 25922 | 16 | <6.2 × 10−10 |
| K. pneumoniae 13883 | 32 | <8.2 × 10−10 |
| A. baumannii 19606 | 32 | <1.3 × 10−9 |
| P. aeruginosa 27853 | 64 | <1.0 × 10−9 |
| Compound | MreB | IC50 (μM) |
|---|---|---|
| TXH11106 | WT | 14 ± 2 |
| CBR-4830 | WT | 49 ± 8 |
| A22 | WT | 447 ± 87 |
| TXH11106 | E143G | >300 |
| Sample | MreB | Km (μM) | Vmax × 102 (μM/s) | kcat × 102 (s−1) | kcat/Km × 104 (μM−1s−1) |
|---|---|---|---|---|---|
| Vehicle | WT | 147 ± 4 | 9.66 ± 0.10 | 4.83 ± 0.05 | 3.29 ± 0.11 |
| TXH11106 | WT | 143 ± 21 | 4.46 ± 0.28 | 2.23 ± 0.14 | 1.56 ± 0.33 |
| CBR-4830 | WT | 150 ± 4 | 6.05 ± 0.07 | 3.03 ± 0.04 | 2.02 ± 0.08 |
| A22 | WT | 151 ± 8 | 8.15 ± 0.19 | 4.08 ± 0.10 | 2.70 ± 0.21 |
| Vehicle | E143G | 170 ± 18 | 0.84 ± 0.04 | 0.42 ± 0.02 | 0.25 ± 0.04 |
| TXH11106 | E143G | 127 ± 28 | 0.76 ± 0.06 | 0.38 ± 0.03 | 0.30 ± 0.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryan, E.J.; Sagong, H.Y.; Parhi, A.K.; Grier, M.C.; Roberge, J.Y.; LaVoie, E.J.; Pilch, D.S. TXH11106: A Third-Generation MreB Inhibitor with Enhanced Activity against a Broad Range of Gram-Negative Bacterial Pathogens. Antibiotics 2022, 11, 693. https://doi.org/10.3390/antibiotics11050693
Bryan EJ, Sagong HY, Parhi AK, Grier MC, Roberge JY, LaVoie EJ, Pilch DS. TXH11106: A Third-Generation MreB Inhibitor with Enhanced Activity against a Broad Range of Gram-Negative Bacterial Pathogens. Antibiotics. 2022; 11(5):693. https://doi.org/10.3390/antibiotics11050693
Chicago/Turabian StyleBryan, Eric J., Hye Yeon Sagong, Ajit K. Parhi, Mark C. Grier, Jacques Y. Roberge, Edmond J. LaVoie, and Daniel S. Pilch. 2022. "TXH11106: A Third-Generation MreB Inhibitor with Enhanced Activity against a Broad Range of Gram-Negative Bacterial Pathogens" Antibiotics 11, no. 5: 693. https://doi.org/10.3390/antibiotics11050693