
Local and Global Protein Interactions Contribute to Residue Entrenchment in Beta-Lactamase TEM-1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
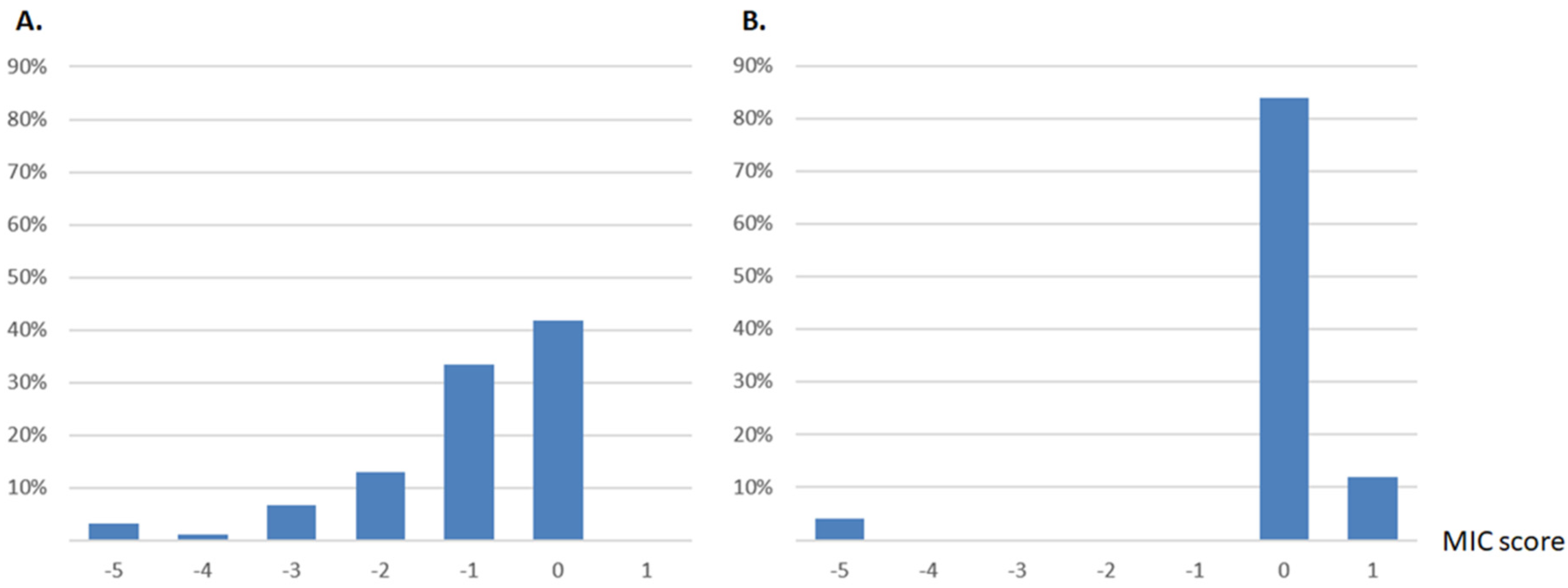
2.1. Distribution of Single Mutant MICs of Two Beta-Lactamases Reveals a Single Opposite Effect
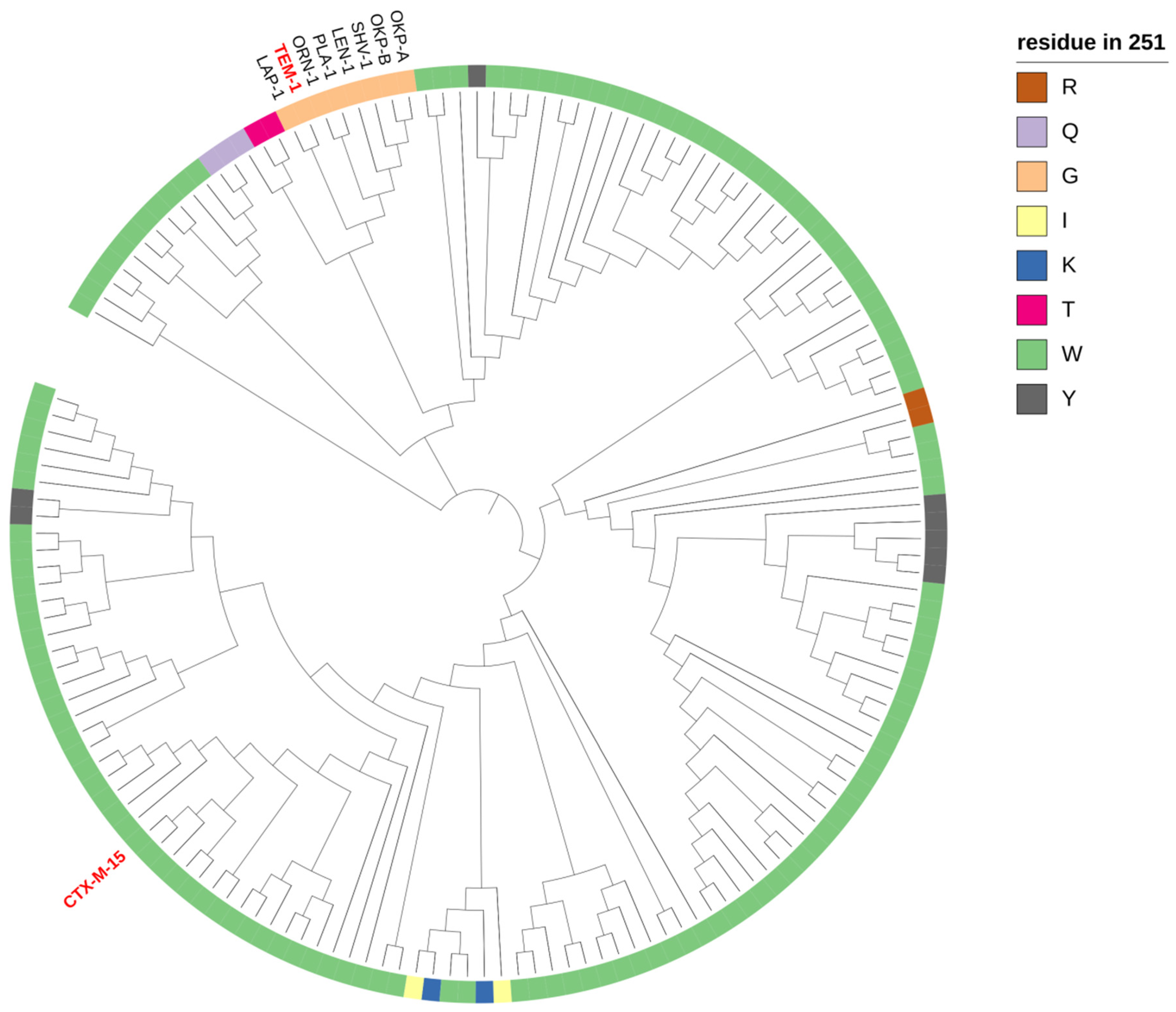
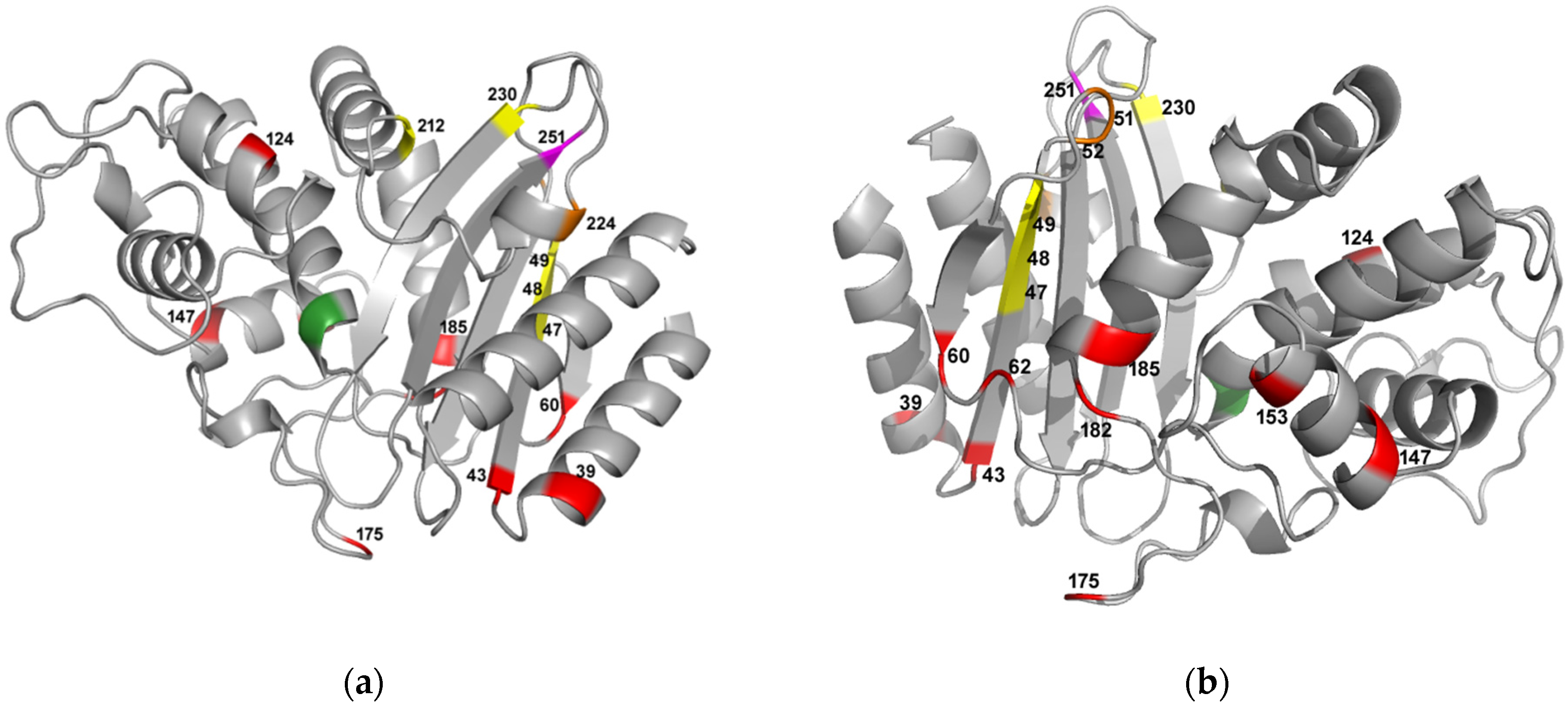
2.2. Phylogeny and Local Environment of Site 251 Highlights a Potential Entrenched Site
2.3. Analysis of Conserved Sites Suggests Local Co-Evolution
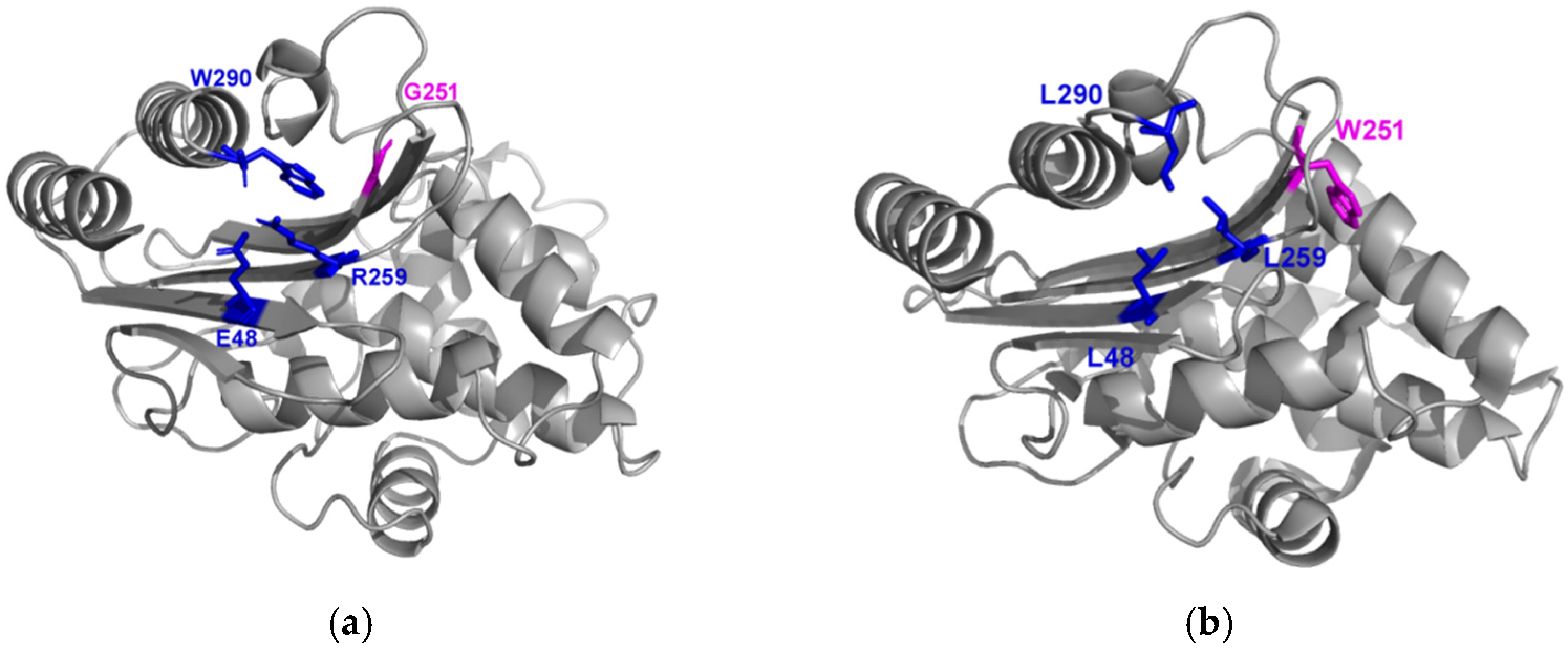
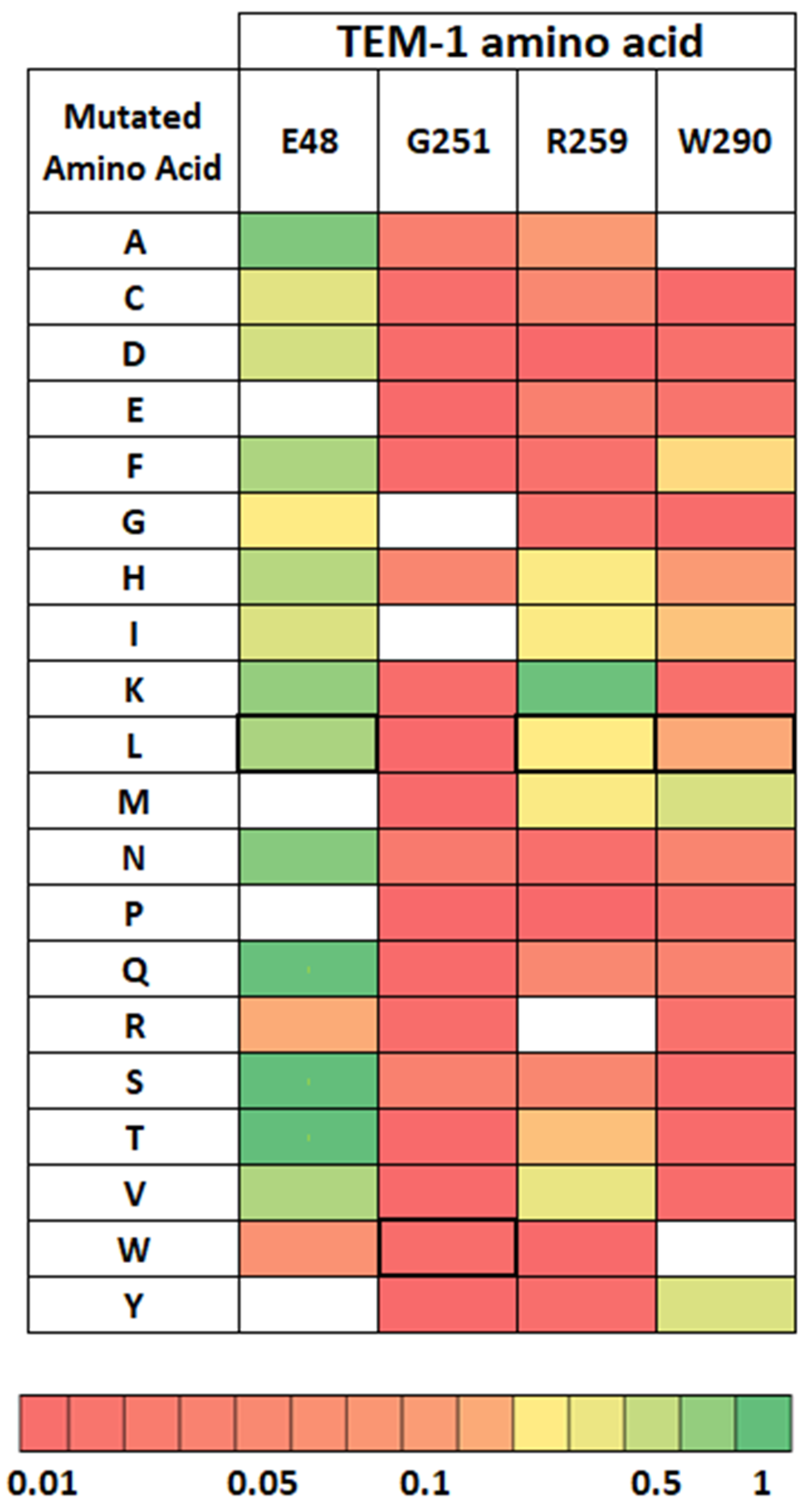
2.4. Distribution of Compensating Mutations in TEM G251W Displays a Various Pattern of Epistatic Interactions
2.4.1. Global Versus Local Epistasis
2.4.2. Mutations with Impact on Global Stability of the Protein
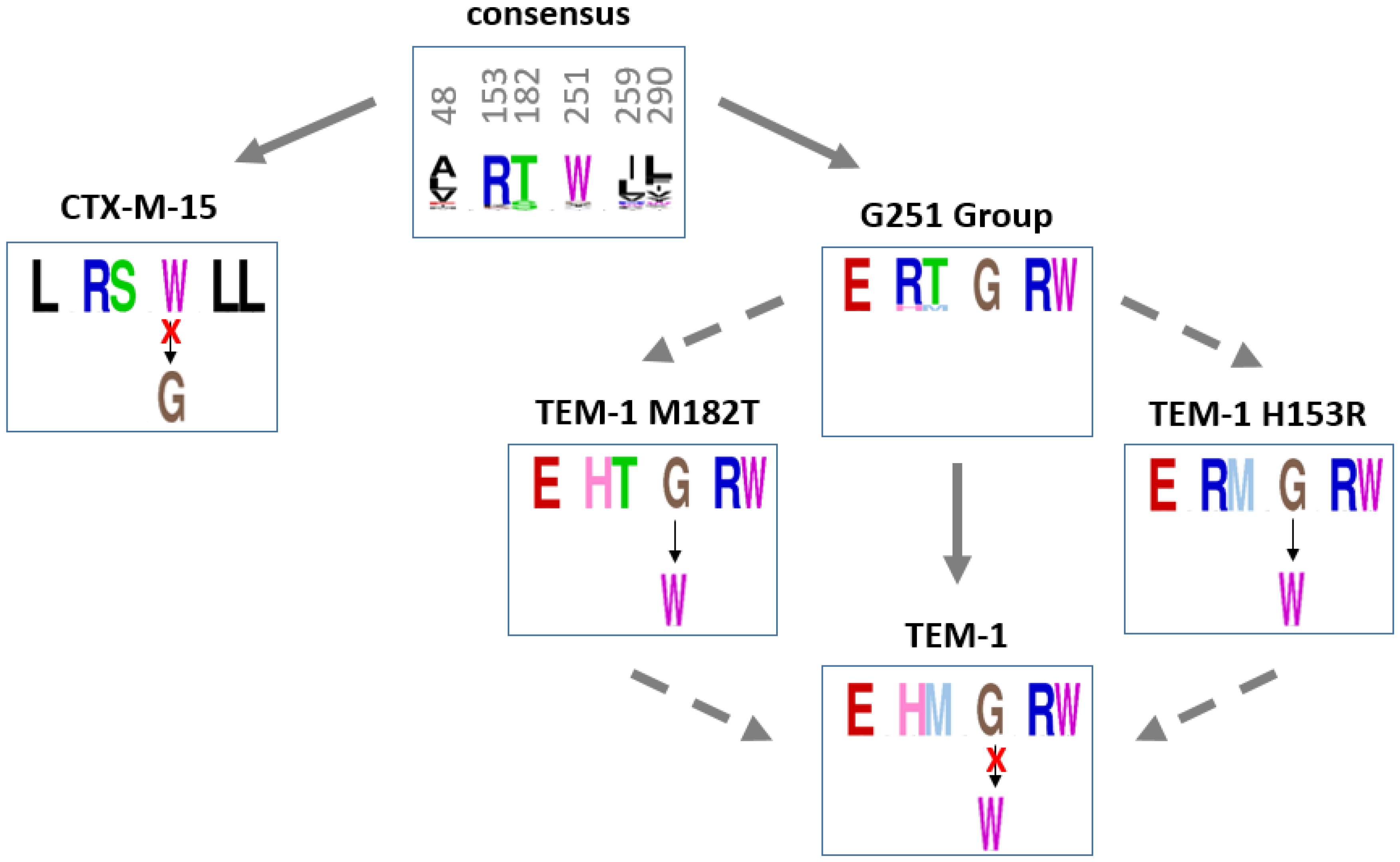
2.4.3. Recovery Mutations in TEM-1 and Consensus Sequences of Beta-Lactamases
2.4.4. Steps of Entrenchment of the Residue G251 in TEM-1
3. Discussion
4. Materials and Methods
4.1. Distribution of Mutation Effects of TEM-1
4.2. Distribution of Mutation Effects of CTX-M-15
CTX-M-15 Library Construction
4.3. MIC Measurements
MIC Score
4.4. Strains
4.5. Plasmids
4.6. Pfunkel Mutagenesis
4.6.1. ssDNA Production
4.6.2. Single Step Pfunkel Mutagenesis
4.6.3. Comprehensive Mutagenesis Pfunkel
4.7. Selection Experiments
4.8. Library Preparation and Deep Sequencing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hall, B.G.; Barlow, M. Evolution of the Serine Beta-Lactamases: Past, Present and Future. Drug Resist. Updates 2004, 7, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Salverda, M.L.M.; De Visser, J.A.G.M.; Barlow, M. Natural Evolution of TEM-1 β-Lactamase: Experimental Reconstruction and Clinical Relevance. FEMS Microbiol. Rev. 2010, 34, 1015–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figliuzzi, M.; Jacquier, H.; Schug, A.; Tenaillon, O.; Weigt, M. Coevolutionary Landscape Inference and the Context-Dependence of Mutations in Beta-Lactamase TEM-1. Mol. Biol. Evol. 2016, 33, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.I.; Suchard, M.A.; Bloom, J.D. Stability-Mediated Epistasis Constrains the Evolution of an Influenza Protein. Elife 2013, 2, e00631. [Google Scholar] [CrossRef]
- Kondrashov, A.S.; Sunyaev, S.; Kondrashov, F.A. Dobzhansky-Muller Incompatibilities in Protein Evolution. Proc. Natl. Acad. Sci. USA 2002, 99, 14878–14883. [Google Scholar] [CrossRef] [Green Version]
- Jordan, D.M.; Frangakis, S.G.; Golzio, C.; Cassa, C.A.; Kurtzberg, J.; Task Force for Neonatal Genomics; Davis, E.E.; Sunyaev, S.R.; Katsanis, N. Identification of Cis-Suppression of Human Disease Mutations by Comparative Genomics. Nature 2015, 524, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.; McCandlish, D.M.; Plotkin, J.B. Contingency and Entrenchment in Protein Evolution under Purifying Selection. Proc. Natl. Acad. Sci. USA 2015, 112, E3226–E3235. [Google Scholar] [CrossRef] [Green Version]
- Firnberg, E.; Labonte, J.W.; Gray, J.J.; Ostermeier, M. A Comprehensive, High-Resolution Map of a Gene’s Fitness Landscape. Mol. Biol. Evol. 2014, 31, 1581–1592. [Google Scholar] [CrossRef] [Green Version]
- Marciano, D.C.; Brown, N.G.; Palzkill, T. Analysis of the Plasticity of Location of the Arg244 Positive Charge within the Active Site of the TEM-1 Beta-Lactamase. Protein Sci. 2009, 18, 2080–2089. [Google Scholar] [CrossRef] [Green Version]
- Naas, T.; Oueslati, S.; Bonnin, R.A.; Dabos, M.L.; Zavala, A.; Dortet, L.; Retailleau, P.; Iorga, B.I. Beta-Lactamase Database (BLDB)—Structure and Function. J. Enzym. Inhib. Med. Chem. 2017, 32, 917–919. [Google Scholar] [CrossRef]
- Caporale, B.; Franceschini, N.; Perilli, M.; Segatore, B.; Rossolini, G.M.; Amicosante, G. Biochemical Characterization of Laboratory Mutants of Extended-Spectrum Beta-Lactamase TEM-60. Antimicrob. Agents Chemother. 2004, 48, 3579–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuna, J.; Pérez-Blancas, A.; Soberón, X. Improving a Circularly Permuted TEM-1 Beta-Lactamase by Directed Evolution. Protein Eng. 2002, 15, 463–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecky, J.; Müller, K.M. Structural Perturbation and Compensation by Directed Evolution at Physiological Temperature Leads to Thermostabilization of Beta-Lactamase. Biochemistry 2005, 44, 12640–12654. [Google Scholar] [CrossRef] [PubMed]
- Bershtein, S.; Goldin, K.; Tawfik, D.S. Intense Neutral Drifts Yield Robust and Evolvable Consensus Proteins. J. Mol. Biol. 2008, 379, 1029–1044. [Google Scholar] [CrossRef] [PubMed]
- Kather, I.; Jakob, R.P.; Dobbek, H.; Schmid, F.X. Increased Folding Stability of TEM-1 Beta-Lactamase by in Vitro Selection. J. Mol. Biol. 2008, 383, 238–251. [Google Scholar] [CrossRef]
- Jacquier, H.; Birgy, A.; Le Nagard, H.; Mechulam, Y.; Schmitt, E.; Glodt, J.; Bercot, B.; Petit, E.; Poulain, J.; Barnaud, G.; et al. Capturing the Mutational Landscape of the Beta-Lactamase TEM-1. Proc. Natl. Acad. Sci. USA 2013, 110, 13067–13072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Palzkill, T. A Natural Polymorphism in Beta-Lactamase Is a Global Suppressor. Proc. Natl. Acad. Sci. USA 1997, 94, 8801–8806. [Google Scholar] [CrossRef] [Green Version]
- Bloom, J.D.; Silberg, J.J.; Wilke, C.O.; Drummond, D.A.; Adami, C.; Arnold, F.H. Thermodynamic Prediction of Protein Neutrality. Proc. Natl. Acad. Sci. USA 2005, 102, 606–611. [Google Scholar] [CrossRef] [Green Version]
- Barlow, M.; Hall, B.G. Predicting Evolutionary Potential: In Vitro Evolution Accurately Reproduces Natural Evolution of the Tem Beta-Lactamase. Genetics 2002, 160, 823–832. [Google Scholar] [CrossRef]
- DePristo, M.A.; Weinreich, D.M.; Hartl, D.L. Missense Meanderings in Sequence Space: A Biophysical View of Protein Evolution. Nat. Rev. Genet. 2005, 6, 678–687. [Google Scholar] [CrossRef]
- Wylie, C.S.; Shakhnovich, E.I. A Biophysical Protein Folding Model Accounts for Most Mutational Fitness Effects in Viruses. Proc. Natl. Acad. Sci. USA 2011, 108, 9916–9921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firnberg, E.; Ostermeier, M. PFunkel: Efficient, Expansive, User-Defined Mutagenesis. PLoS ONE 2012, 7, e52031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalsky, C.A.; Klesmith, J.R.; Stapleton, J.A.; Kelly, V.; Reichkitzer, N.; Whitehead, T.A. High-Resolution Sequence-Function Mapping of Full-Length Proteins. PLoS ONE 2015, 10, e0118193. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazquez, J.; Morosini, M.I.; Negri, M.C.; Gonzalez-Leiza, M.; Baquero, F. Single Amino Acid Replacements at Positions Altered in Naturally Occurring Extended-Spectrum TEM Beta-Lactamases. Antimicrob. Agents Chemother. 1995, 39, 145–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stemmer, W.P. Rapid Evolution of a Protein in Vitro by DNA Shuffling. Nature 1994, 370, 389–391. [Google Scholar] [CrossRef]
- Ortlund, E.A.; Bridgham, J.T.; Redinbo, M.R.; Thornton, J.W. Crystal Structure of an Ancient Protein: Evolution by Conformational Epistasis. Science 2007, 317, 1544–1548. [Google Scholar] [CrossRef] [Green Version]
- Sideraki, V.; Huang, W.; Palzkill, T.; Gilbert, H.F. A Secondary Drug Resistance Mutation of TEM-1 Beta-Lactamase That Suppresses Misfolding and Aggregation. Proc. Natl. Acad. Sci. USA 2001, 98, 283–288. [Google Scholar] [CrossRef]
- Chaïbi, E.B.; Sirot, D.; Paul, G.; Labia, R. Inhibitor-Resistant TEM Beta-Lactamases: Phenotypic, Genetic and Biochemical Characteristics. J. Antimicrob. Chemother. 1999, 43, 447–458. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birgy, A.; Magnan, M.; Hobson, C.A.; Figliuzzi, M.; Panigoni, K.; Codde, C.; Tenaillon, O.; Jacquier, H. Local and Global Protein Interactions Contribute to Residue Entrenchment in Beta-Lactamase TEM-1. Antibiotics 2022, 11, 652. https://doi.org/10.3390/antibiotics11050652
Birgy A, Magnan M, Hobson CA, Figliuzzi M, Panigoni K, Codde C, Tenaillon O, Jacquier H. Local and Global Protein Interactions Contribute to Residue Entrenchment in Beta-Lactamase TEM-1. Antibiotics. 2022; 11(5):652. https://doi.org/10.3390/antibiotics11050652
Chicago/Turabian StyleBirgy, André, Mélanie Magnan, Claire Amaris Hobson, Matteo Figliuzzi, Karine Panigoni, Cyrielle Codde, Olivier Tenaillon, and Hervé Jacquier. 2022. "Local and Global Protein Interactions Contribute to Residue Entrenchment in Beta-Lactamase TEM-1" Antibiotics 11, no. 5: 652. https://doi.org/10.3390/antibiotics11050652