
Streptozotocin-Induced Hyperglycemia Is Associated with Unique Microbiome Metabolomic Signatures in Response to Ciprofloxacin Treatment


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
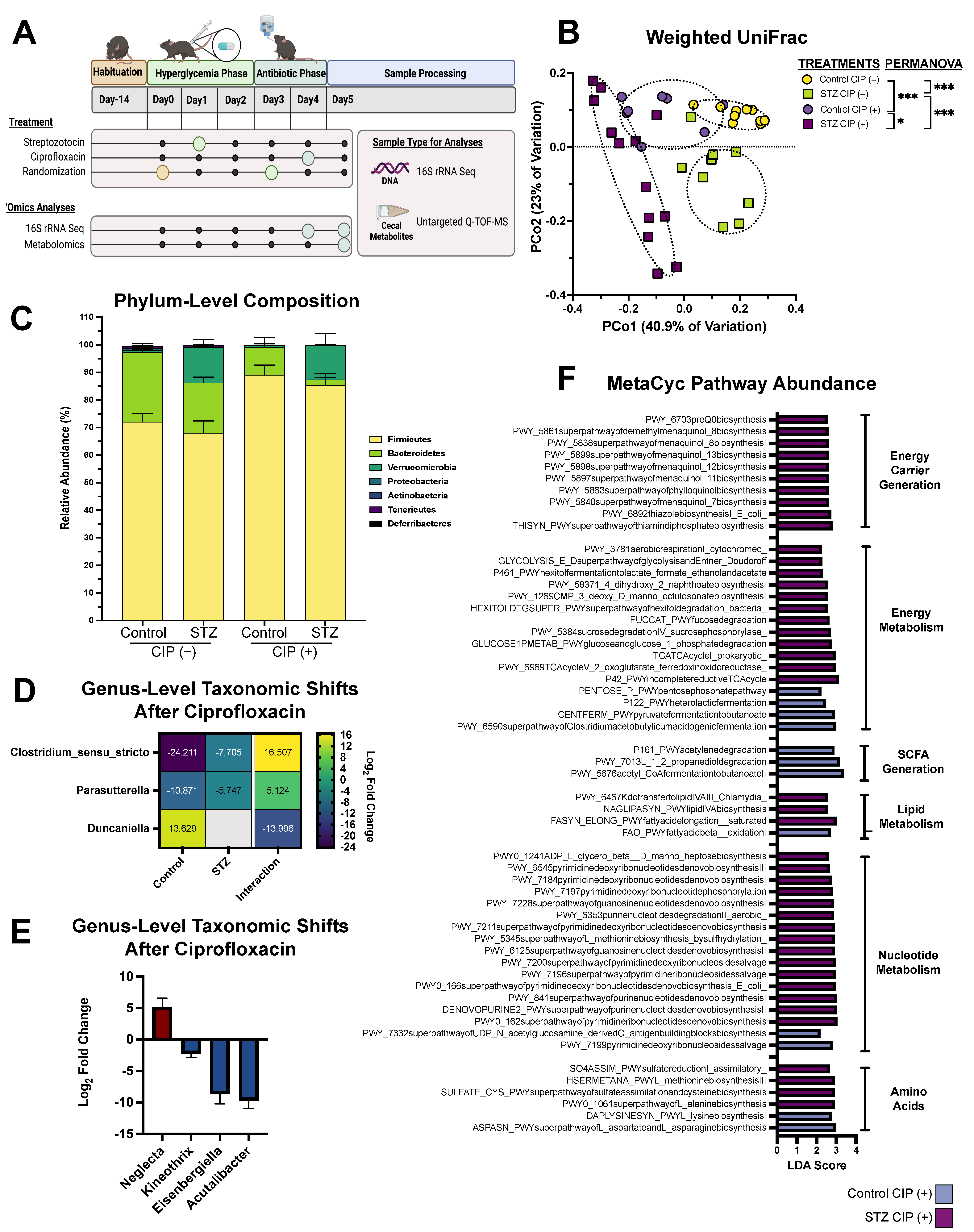
2.1. Animal Experiments
2.2. 16S rRNA Amplicon Sequencing: Library Generation
2.3. 16S rRNA Amplicon Sequencing: Read Processing and Analysis
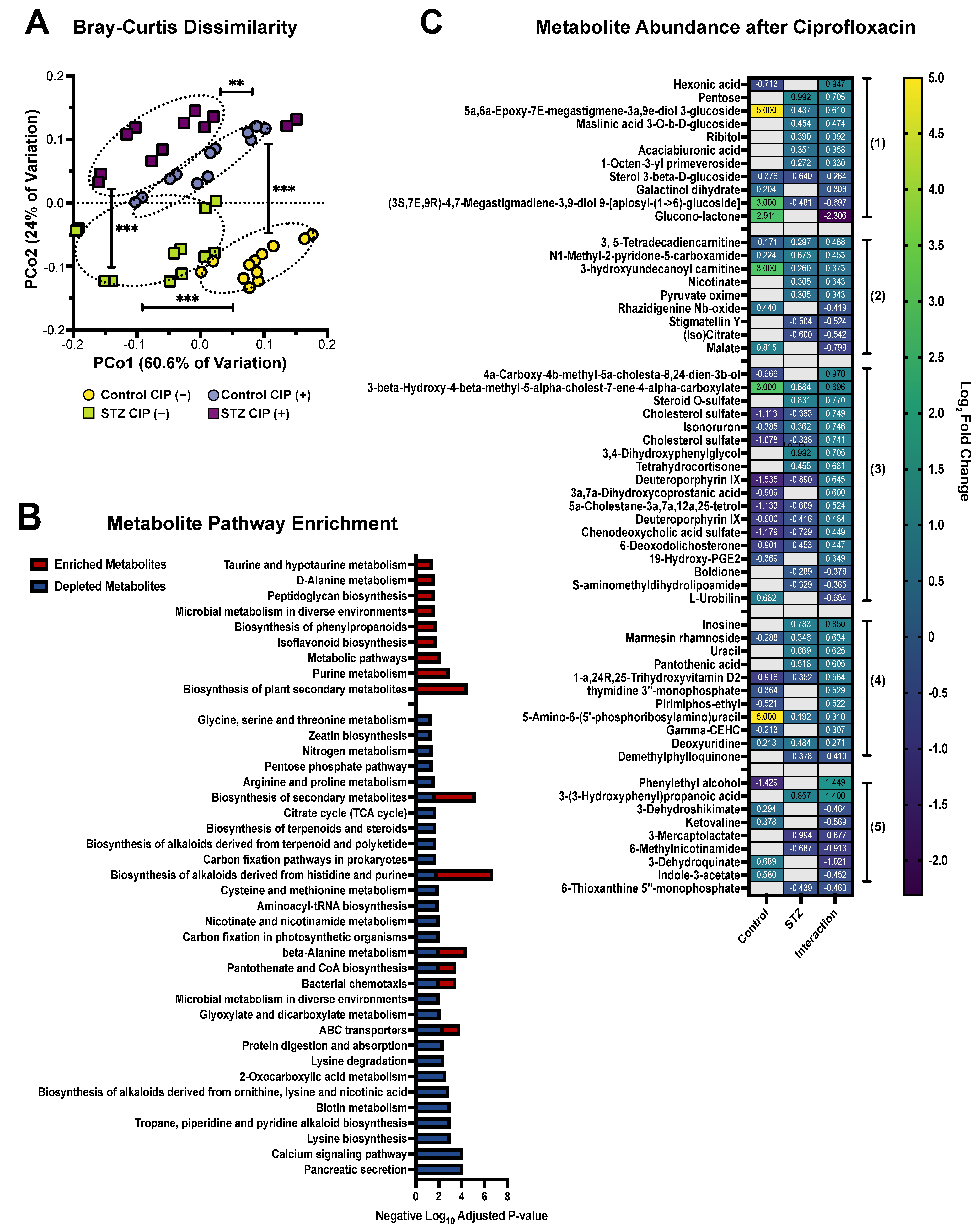
2.4. Q-TOF-MS: Metabolite Extraction and Annotation
2.5. Q-TOF-MS: Computational Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Blaser, M. Antibiotic overuse: Stop the killing of beneficial bacteria. Nature 2011, 476, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108, 4551–4561. [Google Scholar] [CrossRef] [Green Version]
- Modi, S.R.; Lee, H.H.; Spina, C.S.; Collins, J.J. Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature 2013, 499, 219–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral, D.J.; Penumutchu, S.; Reinhart, E.M.; Zhang, C.; Korry, B.J.; Wurster, J.I.; Nilson, R.; Guang, A.; Sano, W.H.; Rowan-Nash, A.D.; et al. Microbial Metabolism Modulates Antibiotic Susceptibility within the Murine Gut Microbiome. Cell Metab. 2019, 30, 800–823.e7. [Google Scholar] [CrossRef] [PubMed]
- Wurster, J.I.; Peterson, R.L.; Brown, C.E.; Penumutchu, S.; Guzior, D.V.; Neugebauer, K.; Sano, W.H.; Sebastian, M.M.; Quinn, R.A.; Belenky, P. Streptozotocin-Induced Hyperglycemia Alters the Cecal Metabolome and Exacerbates Antibiotic Induced Dysbiosis. Cell Rep. 2021, 37, 110113. [Google Scholar] [CrossRef]
- Ni, J.; Friedman, H.; Boyd, B.C.; McGurn, A.; Babinski, P.; Markossian, T.; Dugas, L.R. Early antibiotic exposure and development of asthma and allergic rhinitis in childhood. BMC Pediatr. 2019, 19, 225–228. [Google Scholar] [CrossRef]
- Yoon, M.Y.; Yoon, S.S. Disruption of the Gut Ecosystem by Antibiotics. Yonsei Med. J. 2018, 59, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.M.; Lopatkin, A.J.; Lobritz, M.A.; Collins, J.J. Bacterial Metabolism and Antibiotic Efficacy. Cell Metab. 2019, 30, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Lopatkin, A.J.; Stokes, J.M.; Zheng, E.J.; Yang, J.H.; Takahashi, M.K.; You, L.; Collins, J.J. Bacterial metabolic state more accurately predicts antibiotic lethality than growth rate. Nat. Microbiol. 2019, 4, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Cabral, D.J.; Wurster, J.I.; Belenky, P. Antibiotic Persistence as a Metabolic Adaptation: Stress, Metabolism, the Host, and New Directions. Pharmaceuticals 2018, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.J.; Wang, S.; Meredith, H.R.; Zhuang, B.; Dai, Z.; You, L. Robust, linear correlations between growth rates and β-lactam–mediated lysis rates. Proc. Natl. Acad. Sci. USA 2018, 115, 4069–4074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meylan, S.; Porter, C.B.M.; Yang, J.H.; Belenky, P.; Gutierrez, A.; Lobritz, M.A.; Park, J.; Kim, S.H.; Moskowitz, S.M.; Collins, J.J. Carbon Sources Tune Antibiotic Susceptibility in Pseudomonas aeruginosa via Tricarboxylic Acid Cycle Control. Cell Chem. Biol. 2017, 24, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Jung, J.; Jang, I.-A.; Madsen, E.L.; Park, W. Role of Glyoxylate Shunt in Oxidative Stress Response. J. Biol. Chem. 2016, 291, 11928–11938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolfsen, K.J.; Brynildsen, M.P. Futile cycling increases sensitivity toward oxidative stress in Escherichia coli. Metab. Eng. 2015, 29, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Belenky, P.; Ye, J.D.; Porter, C.B.M.; Cohen, N.R.; Lobritz, M.A.; Ferrante, T.; Jain, S.; Korry, B.J.; Schwarz, E.G.; Walker, G.C.; et al. Bactericidal Antibiotics Induce Toxic Metabolic Perturbations that Lead to Cellular Damage. Cell Rep. 2015, 13, 968–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobritz, M.A.; Belenky, P.; Porter, C.B.M.; Gutierrez, A.; Yang, J.H.; Schwarz, E.G.; Dwyer, D.J.; Khalil, A.S.; Collins, J.J. Antibiotic efficacy is linked to bacterial cellular respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 8173–8180. [Google Scholar] [CrossRef] [Green Version]
- Mok, W.W.K.; Park, J.O.; Rabinowitz, J.D.; Brynildsen, M.P. RNA Futile Cycling in Model Persisters Derived from MazF Accumulation. mBio 2015, 6, e01588-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, V.C.; Kinkead, L.C.; Janssen, A.; Schaeffer, C.R.; Woods, K.M.; Lindgren, J.K.; Peaster, J.M.; Chaudhari, S.S.; Sadykov, M.; Jones, J.; et al. A Dysfunctional Tricarboxylic Acid Cycle Enhances Fitness of Staphylococcus β-Lactam Stress. mBio 2013, 4, e00437-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.; Joshi-Datar, A.; Lepine, F.; Bauerle, E.; Olakanmi, O.; Beer, O.; McKay, G.; Siehnel, R.; Schafhauser, J.; Wang, Y.; et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 2011, 334, 982–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeds, M.C.; Anderson, J.M.; Armstrong, A.S.; Gastineau, D.A.; Hiddinga, H.J.; Jahangir, A.; Eberhardt, N.L.; Kudva, Y.C. Single dose streptozotocin-induced diabetes: Considerations for study design in islet transplantation models. Lab. Anim. 2011, 45, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Barbera, P.; Jozlov, A.M.; Czech, L.; Morel, B.; Darriba, D.; Flouri, T.; Stamatakis, A. EPA-ng: Massively Parallel Evolutionary Placement of Genetic Sequences. Syst. Biol. 2019, 68, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Czech, L.; Barbera, P.; Stamatakis, A. Genesis and Gappa: Processing, analyzing and visualizing phylogenetic (placement) data. Bioinformatics 2020, 36, 685–688. [Google Scholar] [CrossRef] [Green Version]
- Louca, S.; Doubli, M. Efficient comparative phylogenetics on large trees. Bioinformatics 2018, 34, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Doak, T.G. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput. Biol. 2009, 5, e1000465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Fuhrer, T.; Heer, D.; Begemann, B.; Zamboni, N. High-throughput, accurate mass metabolome profiling of cellular extracts by flow injection-time-of-flight mass spectrometry. Anal. Chem. 2011, 83, 7074–7080. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; D’Auria, J.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D.; et al. Mass pectrometry-based metabolomics: A guide for annotation, quantification and best reporting practices. Nat. Meth. 2021, 18, 747–756. [Google Scholar] [CrossRef]
- Aggio, R.B.M.; Ruggiero, K.; Villas-Boas, S.G. Pathway activity profiling (PAPi): From the metabolite profile to the metabolic pathway activity. Bioinformatics 2010, 26, 2969–2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Method. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Cabral, D.J.; Wurster, J.I.; Korry, B.J.; Penumutchu, S.; Belenky, P. Consumption of a Western-Style Diet Modulates the Response of the Murine Gut Microbiome to Ciprofloxacin. mSystems 2020, 5, e00317-20. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-X.; Qin, Y.; Chen, T.; Lu, M.; Qian, X.; Guo, X.; Bai, Y. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 2021, 12, 315–330. [Google Scholar] [CrossRef] [PubMed]
- de Lastours, V.; Fantin, B. Impact of fluoroquinolones on human microbiota. Focus on the emergence of antibiotic resistance. Future Microbiol. 2015, 10, 1241–1255. [Google Scholar] [CrossRef]
- Dorries, K.; Schlueter, R.; Lalk, M. Impact of Antibiotics with Various Target Sites on the Metabolome of Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 7151–7163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falla, T.J.; Chopra, I. Joint tolerance to β-lactam and fluoroquinolone antibiotics in Escherichia coli results from overexpression of hipA. Antimicrob. Agents Chemother. 1998, 42, 3282–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, V.; Sevilla, A.; Cánovas, M.; Iborra, J.L. Production of L-carnitine by secondary metabolism of bacteria. Microb. Cell. Fact. 2007, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meadows, J.A.; Wargo, M.J. Carnitine in bacterial physiology and metabolism. Microbiology 2015, 161, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, C.; Chapman, A.; Fellermeier, F.; Choudhary, M.; Scheel, D.; Glawischnig, E. The Biosynthetic Pathway of Indole-3-Carbaldehyde and Indole-3-Carboxylic Acid Derivatives in Arabidopsis. Plant Physiol. 2014, 165, 841–853. [Google Scholar] [CrossRef] [Green Version]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [Green Version]
- Sachar, M.; Anderson, K.E.; Ma, X. Protoporphyrin IX: The Good, the Bad, and the Ugly. J. Pharmacol. Exper. Ther. 2016, 356, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Jedrey, H.; Lilley, K.S.; Welch, M. Ciprofloxacin binding to GyrA causes global changes in the proteome of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2018, 365, fny134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Chen, X.; Yu, W.; Zhou, B.; Liu, L.; Yang, Y.; Du, P.; Liu, L.; Li, C. Ciprofloxacin stress changes key enzymes and intracellular metabolites of Lactobacillus plantarum DNZ-4. Food Sci. Hum. Wellness 2022, 11, 332–340. [Google Scholar] [CrossRef]
- Lopatkin, A.J.; Bening, S.C.; Manson, A.L.; Stokes, J.M.; Kohanski, M.A.; Badran, A.H.; Earl, A.M.; Cheney, N.J.; Yang, J.H.; Collins, J.J. Clinically relevant mutations in core metabolic genes confer antibiotic resistance. Science 2021, 371, eaba0862. [Google Scholar] [CrossRef]
- Martins, D.; Nguyen, D. Stimulating Central Carbon Metabolism to Re-sensitize Pseudomonas aeruginosa to Aminoglycosides. Cell Chem. Biol. 2017, 24, 122–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertsman, I.; Barshop, B.A. Promises and pitfalls of untargeted metabolomics. J. Inherit. Metab. Dis. 2018, 41, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.A.; Hagiwara, K.A.; Liu, K.; Goudzari, M.; Pathmasiri, W.; Sumner, L.W.; Metz, T.O. Bridging the Gap between Analytical and Microbial Sciences within Microbiome Research. mSystems 2021, 6, e00585-21. [Google Scholar] [CrossRef]
- Fuhrer, T.; Zampieri, M.; Sévin, D.C.; Sauer, U.; Zamboni, N. Genomewide landscape of gene-metabolome associations in Escherichia coli. Mol. Syst. Biol. 2017, 13, 907. [Google Scholar] [CrossRef] [PubMed]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and Limitations of 16S rRNA Gene Amplicon Sequencing in Revealing Temporal Microbial Community Dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wurster, J.I.; Peterson, R.L.; Belenky, P. Streptozotocin-Induced Hyperglycemia Is Associated with Unique Microbiome Metabolomic Signatures in Response to Ciprofloxacin Treatment. Antibiotics 2022, 11, 585. https://doi.org/10.3390/antibiotics11050585
Wurster JI, Peterson RL, Belenky P. Streptozotocin-Induced Hyperglycemia Is Associated with Unique Microbiome Metabolomic Signatures in Response to Ciprofloxacin Treatment. Antibiotics. 2022; 11(5):585. https://doi.org/10.3390/antibiotics11050585
Chicago/Turabian StyleWurster, Jenna I., Rachel L. Peterson, and Peter Belenky. 2022. "Streptozotocin-Induced Hyperglycemia Is Associated with Unique Microbiome Metabolomic Signatures in Response to Ciprofloxacin Treatment" Antibiotics 11, no. 5: 585. https://doi.org/10.3390/antibiotics11050585