
Biocide-Resistant Escherichia coli ST540 Co-Harboring ESBL, dfrA14 Confers QnrS-Dependent Plasmid-Mediated Quinolone Resistance
Abstract
:1. Introduction
2. Results and Discussion
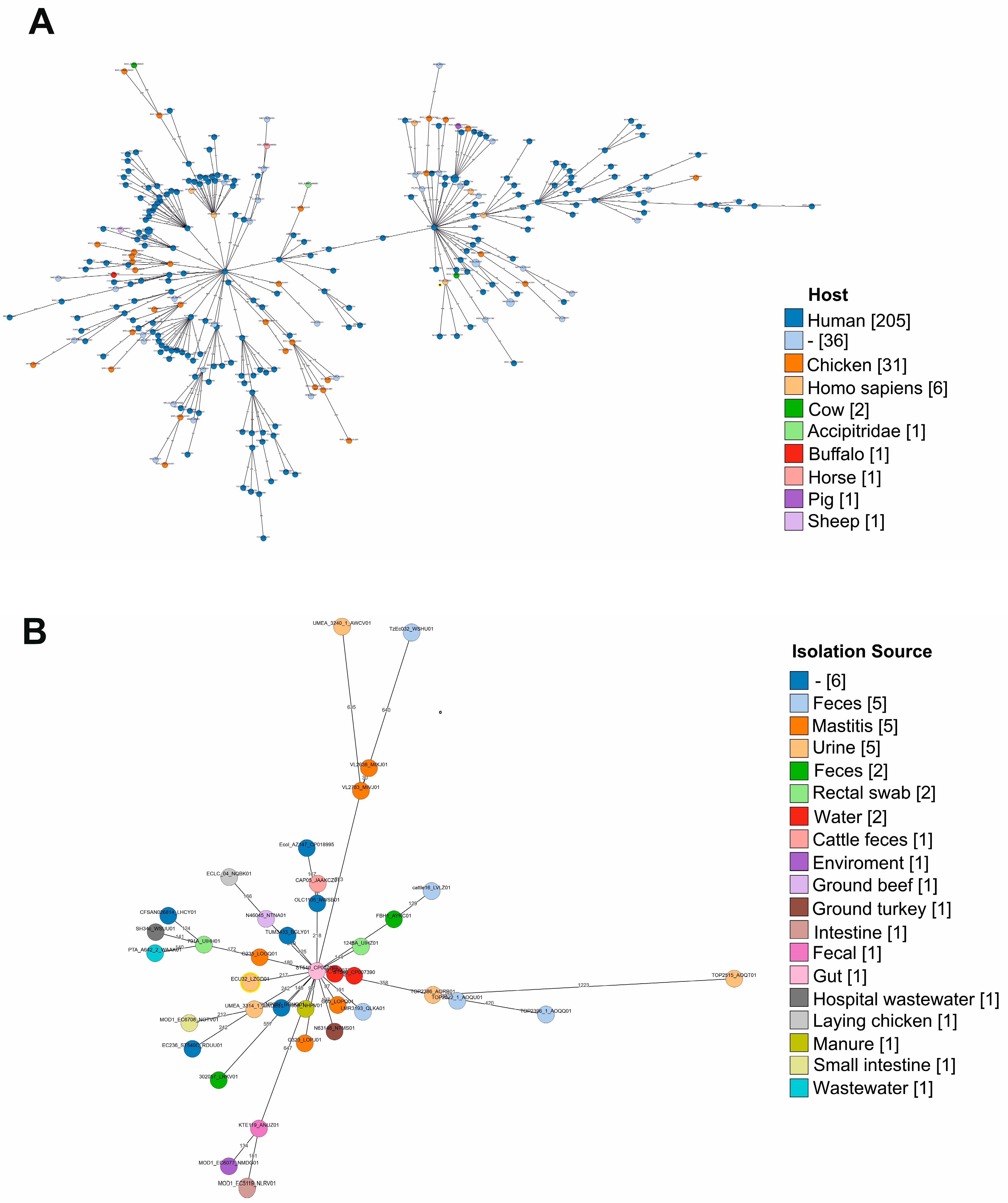
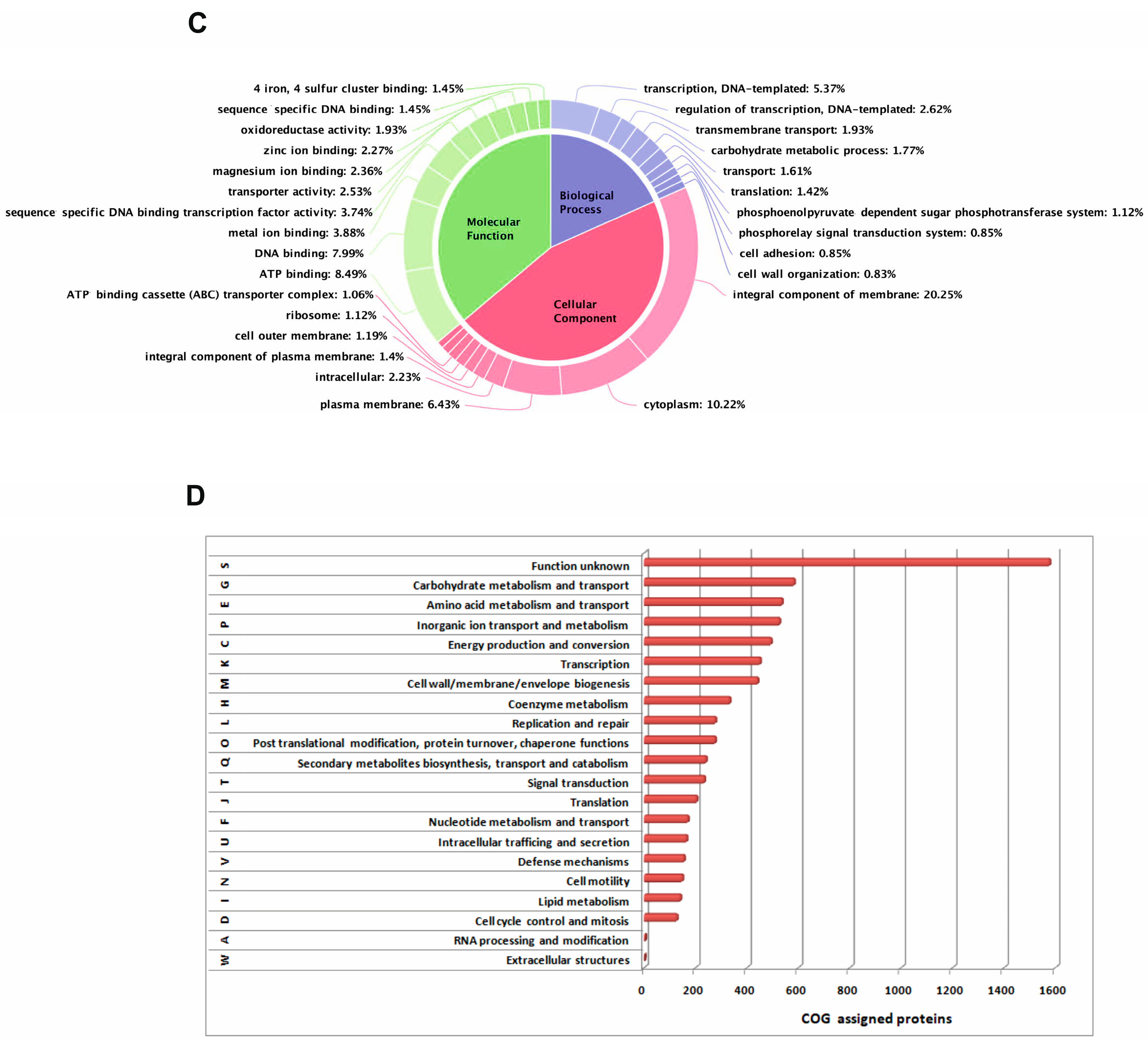
2.1. Genomic Features and Phylogenetic Analysis of E. coli
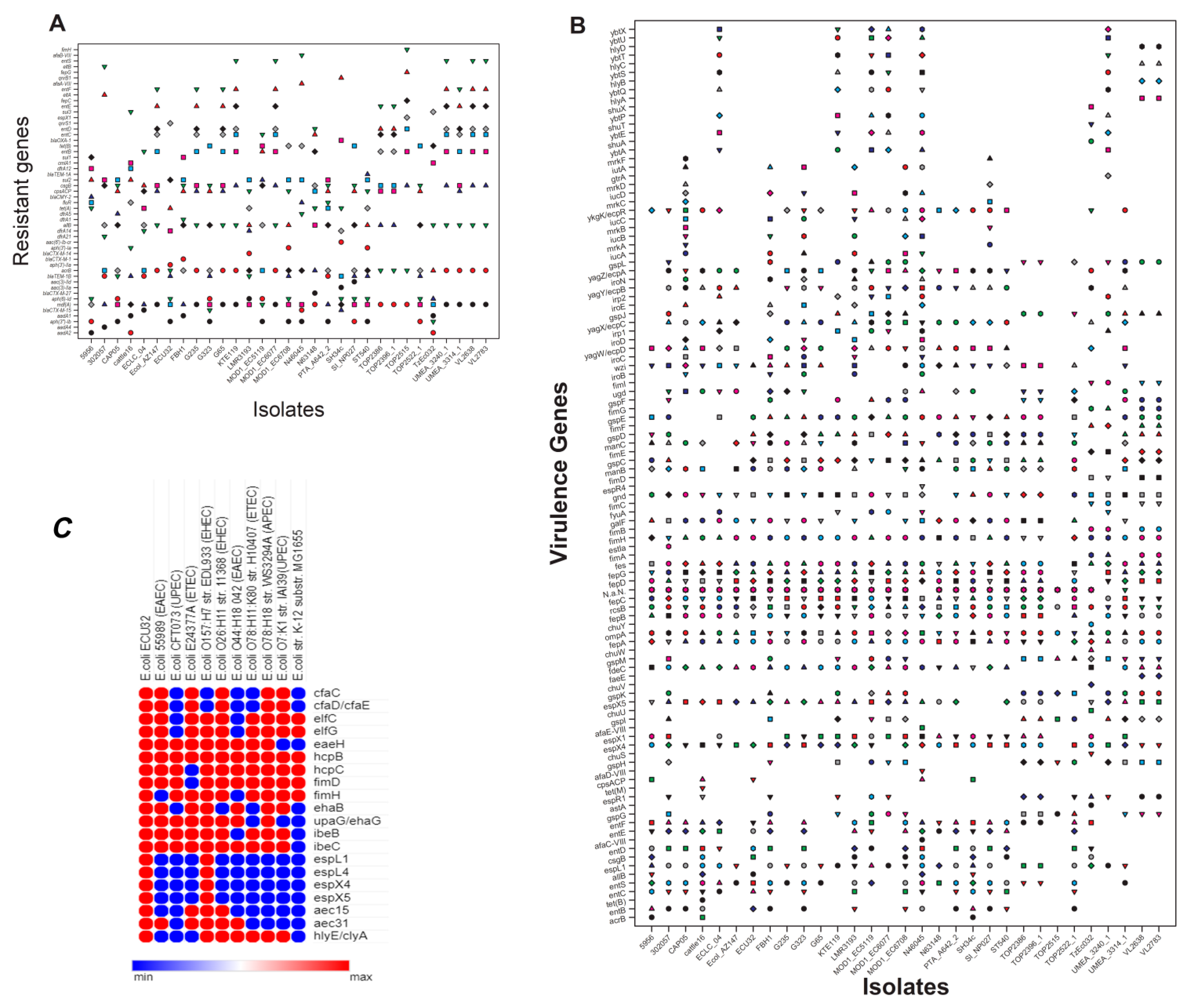
2.2. Antimicrobial-Resistant Genes
2.3. Virulence and Its Associated Genes
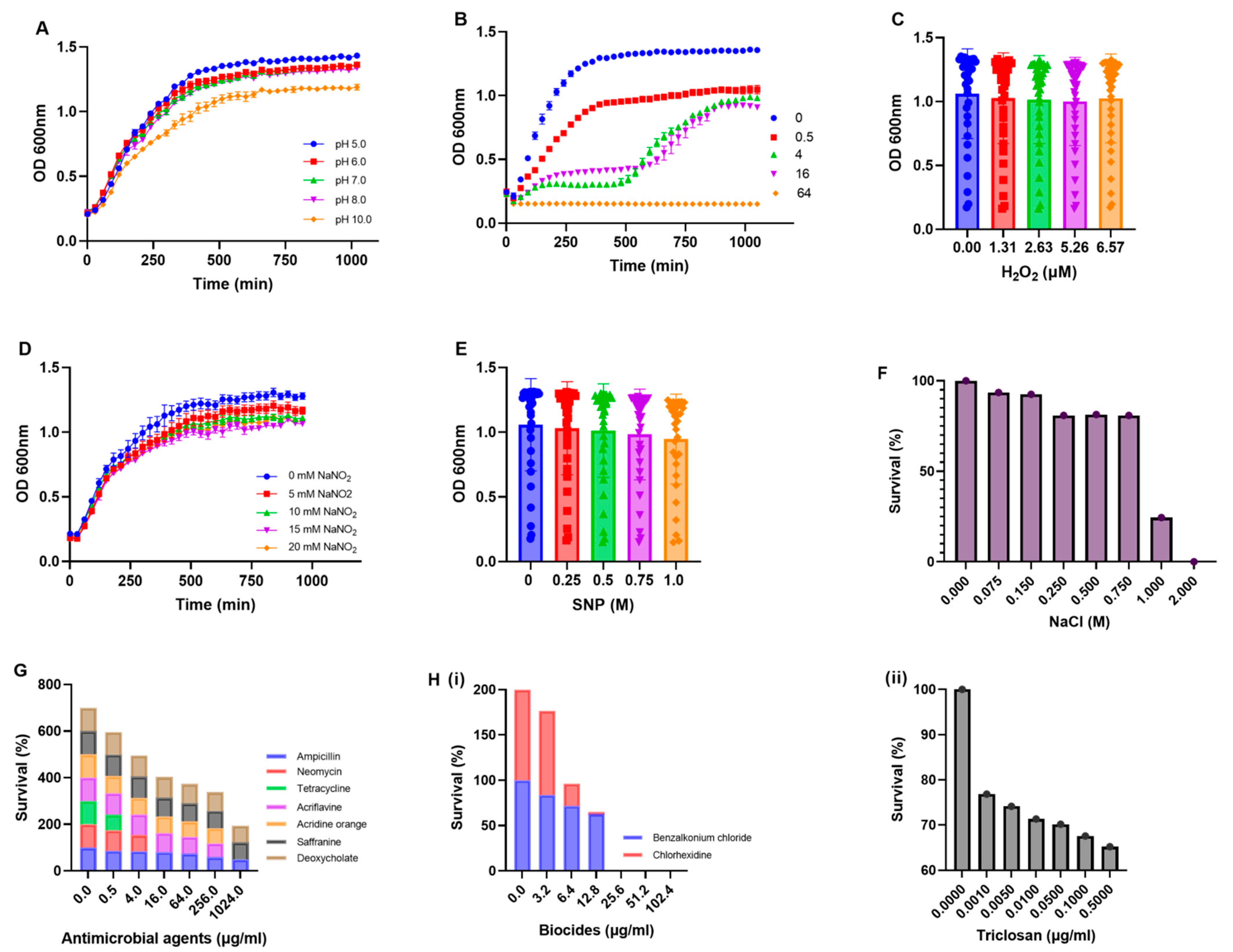
2.4. Phenotypic Characterization of Multidrug-Resistant E. coli

3. Materials and Methods
3.1. The Genome Draft Sequence, Annotation and Analysis
3.2. Antibiotic Resistance Pattern and Growth Analysis
3.3. Data Deposition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Nicolas-Chanoine, M.H.; Bertrand, X.; Madec, J.Y. Escherichia coli ST131, an intriguing clonal group. Clin. Microbiol. Rev. 2014, 3, 543–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, M.; Matsoso, M.P. A global call for action to combat antimicrobial resistance: Can we get it right this time? S. Afr. Med. J. 2014, 7, 478–479. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Denamur, E.; Clermont, O.; Bonacorsi, S.; Gordon, D. The population genetics of pathogenic Escherichia coli. Nat. Rev. Microbiol. 2021, 1, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Croxen, M.A.; Law, R.J.; Scholz, R.; Keeney, K.M.; Wlodarska, M.; Finlay, B.B. Recent advances in understanding enteric pathogenic Escherichia coli. Clin. Microbiol. Rev. 2013, 4, 822–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, A.C.C.; Andrade, N.L.; Ferdous, M.; Chlebowicz, M.A.; Santos, C.C.; Correal, J.C.D.; Lo Ten Foe, J.R.; Rosa, A.C.P.; Damasco, P.V.; Friedrich, A.W.; et al. Comprehensive Molecular Characterization of Escherichia coli Isolates from Urine Samples of Hospitalized Patients in Rio de Janeiro, Brazil. Front. Microbiol. 2018, 9, 243. [Google Scholar] [CrossRef] [PubMed]
- Payne, L.J.; Todeschini, T.C.; Wu, Y.; Perry, B.J.; Ronson, C.W.; Fineran, P.C.; Nobrega, F.L.; Jackson, S.A. Identification and classification of antiviral defence systems in bacteria and archaea with PADLOC reveals new system types. Nucleic Acids Res. 2021, 49, 10868–10878. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.B.; Rajamohan, G. Genome analysis of urease positive Serratia marcescens, co-producing SRT-2 and AAC(6′)-Ic with multidrug efflux pumps for antimicrobial resistance. Genomics 2019, 4, 653–660. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.B.; Rajamohan, G. Comparative genome analysis and characterization of a MDR Klebsiella variicola. Genomics 2020, 5, 3179–3190. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Goh, Y.X.; Tai, C.; Wang, H.; Deng, Z.; Ou, H.Y. VRprofile2: Detection of antibiotic resistance-associated mobilome in bacterial pathogens. Nucleic Acids Res. 2020, 50, W768–W773. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019. A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Assembly Statistics |
|---|---|
| Genome | Escherichia coli ECU32 |
| Size | 4,734,193 |
| GC content | 50.8 |
| Number of coding sequences | 4601 |
| Number of RNAs | 105 |
| Number of subsystems | 595 |
| Contigs generated | 107 |
| Maximum contig length | 239,491 |
| Minimum contig length | 502 |
| Average contig length | 44,244.8 ± 55,998.5 |
| Median contig length | 8912 |
| Total contigs length | 4,734,193 |
| Total number of non-ATGC characters | 663 |
| Percentage of non-ATGC characters | 0.014 |
| Contigs ≥ 500 bp | 107 |
| Contigs ≥ 1 kbp | 97 |
| Contigs ≥ 10 kbp | 67 |
| Contigs ≥ 1 Mbp | 0 |
| N50 value | 95,844 |
| L50 | 15 |
| Genome coverage | 93.0X |
| CRISPR System | Protein | Target | Sequence Id | Start | End | Strand |
|---|---|---|---|---|---|---|
| CRISPR_array | CRISPR_array | CRISPR001 | LZGD01000013.1 | 92,385 | 92,901 | - |
| cas_type_I-E | Cas2e | A8A11_20845 | LZGD01000013.1 | 93,006 | 93,291 | - |
| cas_type_I-E | Cas1e | A8A11_20850 | LZGD01000013.1 | 93,292 | 94,210 | - |
| cas_type_I-E | Cas6e | A8A11_20855 | LZGD01000013.1 | 94,225 | 94,825 | - |
| cas_type_I-E | Cas5e | A8A11_20860 | LZGD01000013.1 | 94,811 | 95,486 | - |
| cas_type_I-E | Cas7e | A8A11_20865 | LZGD01000013.1 | 95,488 | 96,580 | - |
| cas_type_I-E | Cas11e | A8A11_20870 | LZGD01000013.1 | 96,592 | 97,075 | - |
| cas_type_I-E | Cas8e | A8A11_20875 | LZGD01000013.1 | 97,067 | 98,576 | - |
| CRISPR_array | CRISPR_array | CRISPR002 | LZGD01000043.1 | 123,708 | 123,910 | - |
| retron_I-C | RT-Toprim_I-C | A8A11_16640 | LZGD01000043.1 | 170,971 | 172,729 | - |
| retron_I-C | msr-msd | NA | LZGD01000043.1 | 172,765 | 172,895 | - |
| RM_type_II | MTase_II | A8A11_21460 | LZGD01000051.1 | 19,587 | 21,006 | + |
| RM_type_II | REase_II | A8A11_21465 | LZGD01000051.1 | 20,986 | 21,457 | + |
| DMS_other | BrxD | A8A11_09690 | LZGD01000074.1 | 2659 | 3976 | + |
| DMS_other | BrxHI | A8A11_09695 | LZGD01000074.1 | 3972 | 6168 | + |
| CRISPR_array | CRISPR_array | CRISPR003 | LZGD01000087.1 | 154,485 | 155,734 | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bharathi, S.V.; Rajamohan, G. Biocide-Resistant Escherichia coli ST540 Co-Harboring ESBL, dfrA14 Confers QnrS-Dependent Plasmid-Mediated Quinolone Resistance. Antibiotics 2022, 11, 1724. https://doi.org/10.3390/antibiotics11121724
Bharathi SV, Rajamohan G. Biocide-Resistant Escherichia coli ST540 Co-Harboring ESBL, dfrA14 Confers QnrS-Dependent Plasmid-Mediated Quinolone Resistance. Antibiotics. 2022; 11(12):1724. https://doi.org/10.3390/antibiotics11121724
Chicago/Turabian StyleBharathi, Srinivasan Vijaya, and Govindan Rajamohan. 2022. "Biocide-Resistant Escherichia coli ST540 Co-Harboring ESBL, dfrA14 Confers QnrS-Dependent Plasmid-Mediated Quinolone Resistance" Antibiotics 11, no. 12: 1724. https://doi.org/10.3390/antibiotics11121724