
The Issue of Pharmacokinetic-Driven Drug-Drug Interactions of Antibiotics: A Narrative Review

Abstract
:1. Introduction
1.1. Search Strategy
1.2. The Clinical Relevance of pDDIs
1.3. The Mechanisms Underlying pDDIs
1.4. Pharmacokinetic Issues in ICU
2. pDDIs Issues in ICU
3. Antibiotics as Victims or Perpetrators of pDDIs
3.1. Beta-Lactams
3.1.1. Penicillins

{kind=link}
| Study | Study Design | Flucloxacillin Dose | Azole/Dose | Main Findings |
|---|---|---|---|---|
| [28] | Case report | 8 g/day | VRC 400–1000 mg/day | VRC trough fell to <1 mg/L and remained subtherapeutic until flucloxacillin discontinuation |
| [29] | Retrospective, 20 patients | 1–12 g/day | Not reported | 11/20 patients had VRC trough <1 mg/L (median 0.2 mg/L) |
| [30] | Case report 1 | 12 g/day | VRC, 4–8 mg/kg bid | 1st VRC trough: <0.2 mg/L; 2nd VRC trough: <0.2 mg/L; 3rd VRC trough: 3 mg/L (after flucloxacillin discontinuation) |
| [30] | Case report 2 | 12 g/day | ISA 200 mg/day | ISA trough increased from <0.3 mg/L to 1.7–5.2 mg/L after flucloxacillin discontinuation |
| [31] | Case report 1 | 8 g/day | VRC 4–8 mg/kg bid; ISA 200 mg/day | VRC trough: 0.6 mg/L; ISA trough increased from 0.4 to 2 mg/L after flucloxacillin discontinuation |
| [31] | Case report 2 | 12 g/day | VRC 300 mg bid ISA 200 mg/day/bid | VRC trough: <0.2 mg/L; ISA trough increased from 0.6–1.5 mg/L to 2.6–5.1 mg/L after flucloxacillin discontinuation |
| [32] | Retrospective, 33 patients | Not reported | Not reported | VRC trough: 0.5 (0–1.8) mg versus 3.5 (1.7–5.1) mg/L in patients given or not flucloxacillin |
| [33] | Case report | 8 g/day | VRC 200 mg bid; POS 300 mg bid | VRC trough reduced from 2.2 to <0.2 mg/L after adding g; POS trough reduced from 1.4 to 0.8 mg/L after adding flucloxacillin |
3.1.2. Cephalosporins
3.1.3. Monobactams
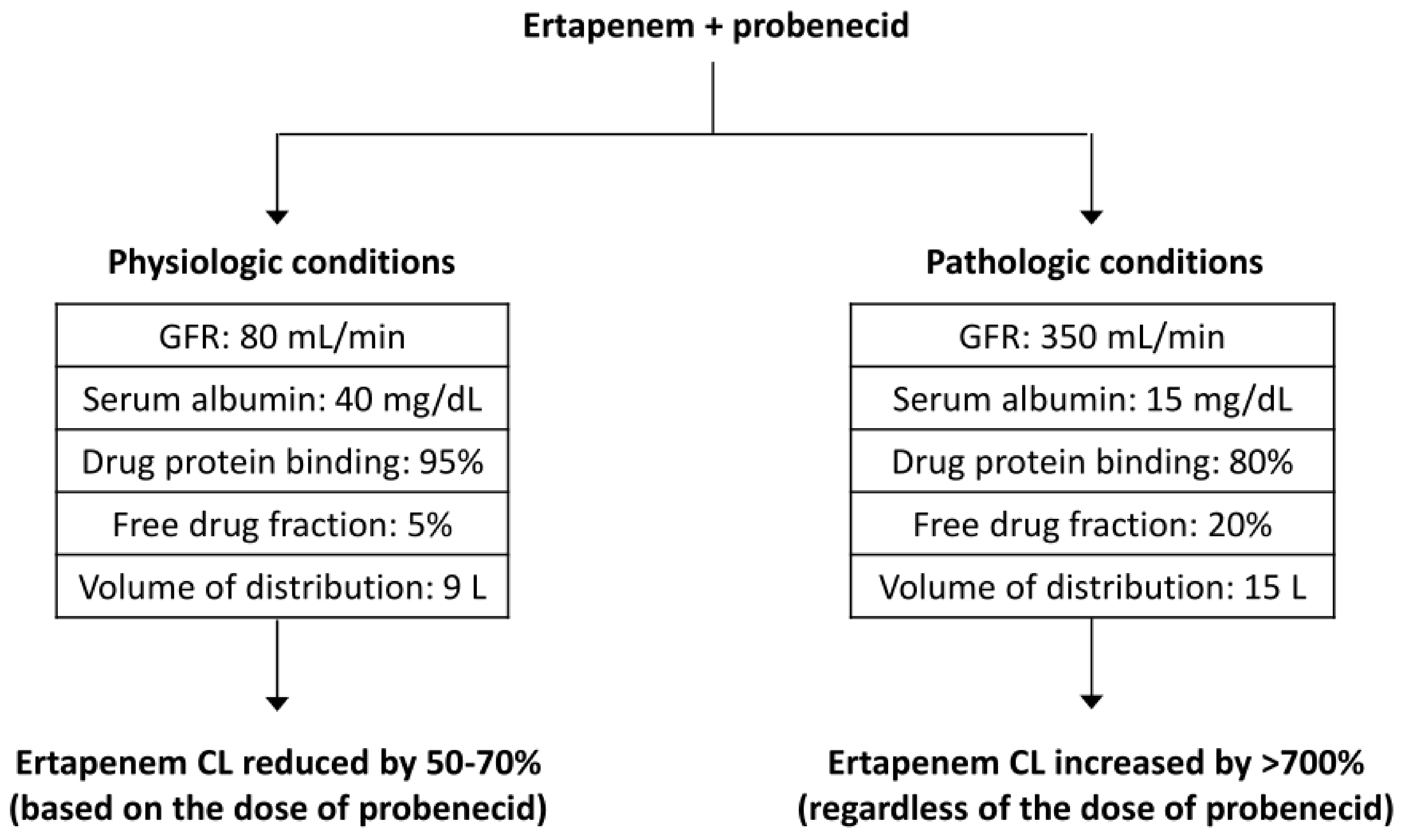
3.1.4. Carbapenems
3.2. Glycopeptides and Lipoglycopeptides
3.3. Tetracyclines
3.4. Macrolides
3.5. Fluoroquinolones
3.5.1. Fluoroquinolones as Victims
3.5.2. Fluoroquinolones as Perpetrators
3.6. Oxazolidinones
3.6.1. Oxazolidinones as Victims of pDDIs
3.6.2. Oxazolidinones as Perpetrators of pDDIs
3.7. Aminoglycosides
3.8. Sulfonamides and Trimethoprim
3.9. Rifamycins
3.9.1. Rifamycins as Victims of DDIs
3.9.2. Rifamycins as Perpetrators of DDIs
4. Tools to Handle pDDIs Involving Antibiotics in ICU
4.1. Drug-Interaction Checkers
4.2. Therapeutic Drug Monitoring
4.3. Physiologically-Based Pharmacokinetic Modelling
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kollef, M.H.; Bassetti, M.; Francois, B.; Burnham, J.; Dimopoulos, G.; Garnacho-Montero, J.; Lipman, J.; Luyt, C.E.; Nicolau, D.P.; Postma, M.J.; et al. The intensive care medicine research agenda on multidrug-resistant bacteria, antibiotics, and stewardship. Intensive Care Med. 2017, 43, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Shankar-Hari, M.; Phillips, G.S.; Levy, M.L.; Seymour, C.W.; Liu, V.X.; Deutschman, C.S.; Angus, D.C.; Rubenfeld, G.D.; Singer, M. Sepsis Definitions Task Force. Developing a New Definition and Assessing New Clinical Criteria for Septic Shock: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 775–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, A.C.; Stevens, J.P.; Walkey, A.J. National Trends in Timing of Death Among Patients with Septic Shock, 1994–2014. Crit. Care Med. 2019, 47, 1493–1496. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, R.D.; Miller, M.; Albertson, T.; Panacek, E.; Johnson, D.; Teoh, L.; Barchuk, W. Adequacy of early empiric antibiotic treatment and survival in severe sepsis: Experience from the MONARCS trial. Clin. Infect. Dis. 2004, 38, 284–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morandi, A.; Vasilevskis, E.; Pandharipande, P.P.; Girard, T.D.; Solberg, L.M.; Neal, E.B.; Koestner, T.; Torres, R.E.; Thompson, J.L.; Shintani, A.K.; et al. Inappropriate medication prescriptions in elderly adults surviving an intensive care unit hospitalization. J. Am. Geriatr. Soc. 2013, 61, 1128–1134. [Google Scholar] [CrossRef] [Green Version]
- Andersen, F.H.; Ariansen Haaland, Ø.; Klepstad, P.; Flaatten, H. Frailty and survival in elderly intensive care patients in Norway. Acta Anaesthesiol. Scand. 2021, 65, 1065–1072. [Google Scholar] [CrossRef]
- Bakker, T.; Dongelmans, D.A.; Nabovati, E.; Eslami, S.; de Keizer, N.F.; Abu-Hanna, A.; Klopotowska, J.E. Heterogeneity in the Identification of Potential Drug-Drug Interactions in the Intensive Care Unit: A Systematic Review, Critical Appraisal, and Reporting Recommendations. J. Clin. Pharmacol. 2022, 62, 706–720. [Google Scholar] [CrossRef]
- Łój, P.; Olender, A.; Ślęzak, W.; Krzych, Ł.J. Pharmacokinetic drug-drug interactions in the intensive care unit—Single-centre experience and literature review. Anaesthesiol. Intensive Ther. 2017, 49, 259–267. [Google Scholar]
- Gervasoni, C.; Formenti, T.; Cattaneo, D. Management of Polypharmacy and Drug-Drug Interactions in HIV Patients: A 2-year Experience of a Multidisciplinary Outpatient Clinic. AIDS Rev. 2019, 21, 40–49. [Google Scholar] [CrossRef]
- Eljaaly, K.; Helal, A.; Almandeel, T.; Algarni, R.; Alshehri, S. Multivalent cations interactions with fluoroquinolones or tetracyclines: A cross-sectional study. Saudi J. Biol. Sci. 2021, 28, 6929–6932. [Google Scholar] [CrossRef]
- Gessner, A.; König, J.; Fromm, M.F. Clinical Aspects of Transporter-Mediated Drug-Drug Interactions. Clin. Pharmacol. Ther. 2019, 105, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Grün, B.; Kiessling, M.K.; Burhenne, J.; Riedel, K.D.; Weiss, J.; Rauch, G.; Haefeli, W.E.; Czock, D. Trimethoprim-metformin interaction and its genetic modulation by OCT2 and MATE1 transporters. Br. J. Clin. Pharmacol. 2013, 76, 787–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedrig, D.; Maechler, S.; Hoppe, L.; Corti, N.; Kovari, H.; Russmann, S. Drug safety of macrolide and quinolone antibiotics in a tertiary care hospital: Administration of interacting co-medication and QT prolongation. Eur. J. Clin. Pharmacol. 2016, 72, 859–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaneo, D.; Corona, A.; De Rosa, F.G.; Gervasoni, C.; Kocic, D.; Marriott, D.J. The management of anti-infective agents in intensive care units: The potential role of a ‘fast’ pharmacology. Expert Rev. Clin. Pharmacol. 2020, 13, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Pea, F. Intracellular Pharmacokinetics of Antibacterials and Their Clinical Implications. Clin. Pharmacokinet. 2018, 57, 177–189. [Google Scholar] [CrossRef]
- Roberts, J.A.; Pea, F.; Lipman, J. The clinical relevance of plasma protein binding changes. Clin. Pharmacokinet. 2013, 52, 1–8. [Google Scholar] [CrossRef]
- Silva, C.M.; Baptista, J.P.; Santos, I.; Martins, P. Recommended Antibiotic Dosage Regimens in Critically Ill Patients with Augmented Renal Clearance: A Systematic Review. Int. J. Antimicrob. Agents 2022, 59, 106569. [Google Scholar] [CrossRef]
- Gatti, M.; Pea, F. Antimicrobial Dose Reduction in Continuous Renal Replacement Therapy: Myth or Real Need? A Practical Approach for Guiding Dose Optimization of Novel Antibiotics. Clin. Pharmacokinet. 2021, 60, 1271–1289. [Google Scholar] [CrossRef]
- Brown, G.R. Cephalosporin-probenecid drug interactions. Clin. Pharmacokinet. 1993, 24, 289–300. [Google Scholar] [CrossRef]
- Kane-Gill, S.L.; Kirisci, L.; Verrico, M.M.; Rothschild, J.M. Analysis of risk factors for adverse drug events in critically ill patients. Crit. Care Med. 2012, 40, 823–828. [Google Scholar] [CrossRef] [Green Version]
- Kuscu, F.; Ulu, A.; Inal, A.S.; Suntur, B.M.; Aydemir, H.; Gul, S.; Ecemis, K.; Komur, S.; Kurtaran, B.; Ozkan Kuscu, O.; et al. Potential Drug-Drug Interactions with Antimicrobials in Hospitalized Patients: A Multicenter Point-Prevalence Study. Med. Sci. Monit. 2018, 24, 4240–4247. [Google Scholar] [CrossRef] [PubMed]
- Mehralian, H.A.; Moghaddasi, J.; Rafiei, H. The prevalence of potentially beneficial and harmful drug-drug interactions in intensive care units. Drug Metab. Pers. Ther. 2019, 34, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Etchegoyen, C.V.; Keller, G.A.; Mrad, S.; Cheng, S.; Di Girolamo, G. Drug-induced QT Interval Prolongation in the Intensive Care Unit. Curr. Clin. Pharmacol. 2017, 12, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E. Interactions between oral contraceptives and antifungals/antibacterials. Is contraceptive failure the result? Clin. Pharmacokinet. 1999, 36, 309–313. [Google Scholar] [CrossRef]
- Balis, F.M. Pharmacokinetic drug interactions of commonly used anticancer drugs. Clin. Pharmacokinet. 1986, 11, 223–235. [Google Scholar] [CrossRef]
- Hayes, A.H., Jr. Therapeutic implications of drug interactions with acetaminophen and aspirin. Arch. Intern. Med. 1981, 141, 301–304. [Google Scholar] [CrossRef]
- Nierenberg, D.W. Drug inhibition of penicillin tubular secretion: Concordance between in vitro and clinical findings. J. Pharmacol. Exp. Ther. 1987, 240, 712–716. [Google Scholar]
- Kennedy, B.; Larcombe, R.; Chaptini, C.; Gordon, D.L. Interaction between voriconazole and flucloxacillin during treatment of disseminated Scedosporium apiospermum infection. J. Antimicrob. Chemother. 2015, 70, 2171–2173. [Google Scholar] [CrossRef] [Green Version]
- Muilwijk, E.W.; Dekkers, B.G.J.; Henriet, S.S.V.; Verweij, P.E.; Witjes, B.; Lashof, A.M.L.O.; Groeneveld, G.H.; van der Hoeven, J.; Alffenaar, J.W.C.; Russel, F.G.M.; et al. Flucloxacillin Results in Suboptimal Plasma Voriconazole Concentrations. Antimicrob. Agents Chemother. 2017, 61, e00915–e00917. [Google Scholar] [CrossRef] [Green Version]
- Vangheluwe, T.; Van Hoecke, F.; Dumoulin, A.; Vogelaers, D. Broad-spectrum azoles and flucloxacillin: A dangerous match. Eur. J. Clin. Microbiol. Infect. Dis. 2022, 41, 153–154. [Google Scholar] [CrossRef]
- Van Daele, R.; Wauters, J.; Vandenbriele, C.; Lagrou, K.; Vos, R.; Debaveye, Y.; Spriet, I. Interaction between flucloxacillin and azoles: Is isavuconazole next? Mycoses 2021, 64, 1508–1511. [Google Scholar] [CrossRef] [PubMed]
- Van Daele, R.; Wauters, J.; De Cock, P.; Buyle, F.; Leys, J.; Van Brantegem, P.; Gijsen, M.; Annaert, P.; Debaveye, Y.; Lagrou, K.; et al. Concomitant Treatment with Voriconazole and Flucloxacillin: A Combination to Avoid. Antibiotics 2021, 10, 1112. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, S.; Godinas, L.; Spriet, I.; Vos, R.; Verleden, G.M. Interaction between posaconazole and flucloxacillin in a lung transplant patient: Decrease in plasma exposure of posaconazole and possible undertreatment of invasive aspergillosis: Case report. BMC Pulm. Med. 2022, 22, 110. [Google Scholar] [CrossRef] [PubMed]
- Murthy, B.; Schmitt-Hoffmann, A. Pharmacokinetics and pharmacodynamics of ceftobiprole, an anti-MRSA cephalosporin with broad-spectrum activity. Clin. Pharmacokinet. 2008, 47, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Kunze, A.; Huwyler, J.; Camenisch, G.; Poller, B. Prediction of organic anion-transporting polypeptide 1B1- and 1B3-mediated hepatic uptake of statins based on transporter protein expression and activity data. Drug Metab. Dispos. 2014, 42, 1514–1521. [Google Scholar] [CrossRef] [Green Version]
- Kiang, T.K.; Wilby, K.J.; Ensom, M.H. A critical review on the clinical pharmacokinetics, pharmacodynamics, and clinical trials of ceftaroline. Clin. Pharmacokinet. 2015, 54, 915–931. [Google Scholar] [CrossRef]
- Bilal, M.; El Tabei, L.; Büsker, S.; Krauss, C.; Fuhr, U.; Taubert, M. Clinical Pharmacokinetics and Pharmacodynamics of Cefiderocol. Clin. Pharmacokinet. 2021, 60, 1495–1508. [Google Scholar] [CrossRef]
- Wagenlehner, F.M.; Alidjanov, J.F. Efficacy, pharmacokinetic and pharmacodynamic profile of ceftolozane + tazobactam in the treatment of complicated urinary tract infections. Expert Opin. Drug Metab. Toxicol. 2016, 12, 959–966. [Google Scholar] [CrossRef]
- Mancl, E.E.; Gidal, B.E. The effect of carbapenem antibiotics on plasma concentrations of valproic acid. Ann. Pharmacother. 2009, 43, 2082–2087. [Google Scholar] [CrossRef]
- Lunde, J.L.; Nelson, R.E.; Storandt, H.F. Acute seizures in a patient receiving divalproex sodium after starting ertapenem therapy. Pharmacotherapy 2007, 27, 1202–1205. [Google Scholar] [CrossRef]
- Fudio, S.; Carcas, A.; Piñana, E.; Ortega, R. Epileptic seizures caused by low valproic acid levels from an interaction with meropenem. J. Clin. Pharm. Ther. 2006, 31, 393–396. [Google Scholar] [CrossRef]
- Santucci, M.; Parmeggiani, A.; Riva, R. Seizure worsening caused by decreased serum valproate during meropenem therapy. J. Child Neurol. 2005, 20, 456–457. [Google Scholar] [CrossRef] [PubMed]
- Coves-Orts, F.J.; Borrás-Blasco, J.; Navarro-Ruiz, A.; Murcia-López, A.; Palacios-Ortega, F. Acute seizures due to a probable interaction between valproic acid and meropenem. Ann. Pharmacother. 2005, 39, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Smolders, E.J.; Ter Heine, R.; Natsch, S.; Kramers, K. Meropenem to Treat Valproic Acid Intoxication. Ther. Drug Monit. 2022, 44, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Dreucean, D.; Beres, K.; McNierney-Moore, A.; Gravino, D. Use of meropenem to treat valproic acid overdose. Am. J. Emerg. Med. 2019, 37, 2120.e5–2120.e7. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Brenner, T.; Hatiboglu, G.; Burhenne, J.; Weiss, J.; Weigand, M.A.; Haefeli, W.E. Substantial Impairment of Voriconazole Clearance by High-Dose Meropenem in a Patient with Renal Failure. Clin. Infect. Dis. 2017, 65, 1033–1036. [Google Scholar] [CrossRef] [Green Version]
- Van Bambeke, F.; Van Laethem, Y.; Courvalin, P.; Tulkens, P.M. Glycopeptide antibiotics: From conventional molecules to new derivatives. Drugs 2004, 64, 913–936. [Google Scholar] [CrossRef]
- Klinker, K.P.; Borgert, S.J. Beyond.Vancomycin: The Tail of the Lipoglycopeptides. Clin. Ther. 2015, 37, 2619–2636. [Google Scholar] [CrossRef]
- Roberts, K.D.; Sulaiman, R.M.; Rybak, M.J. Dalbavancin and Oritavancin: An Innovative Approach to the Treatment of Gram-Positive Infections. Pharmacotherapy 2015, 35, 935–948. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Calic, D.; Schweizer, F.; Zelenitsky, S.; Adam, H.; Lagacé-Wiens, P.R.; Rubinstein, E.; Gin, A.S.; Hoban, D.J.; Karlowsky, J.A. New lipoglycopeptides: A comparative review of dalbavancin, oritavancin and telavancin. Drugs 2010, 70, 859–886. [Google Scholar] [CrossRef]
- Murphy, A.A.; Zacur, H.A.; Charache, P.; Burkman, R.T. The effect of tetracycline on levels of oral contraceptives. Am. J. Obstet. Gynecol. 1991, 164, 28–33. [Google Scholar] [CrossRef]
- Barbour, A.; Schmidt, S.; Ma, B.; Schiefelbein, L.; Rand, K.H.; Burkhardt, O.; Derendorf, H. Clinical pharmacokinetics and pharmacodynamics of tigecycline. Clin. Pharmacokinet. 2009, 48, 575–584. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.W. Clinical Pharmacokinetics and Pharmacodynamics of Eravacycline. Clin. Pharmacokinet. 2019, 58, 1149–1153. [Google Scholar] [CrossRef]
- Rodvold, K.A.; Burgos, R.M.; Tan, X.; Pai, M.P. Omadacycline: A Review of the Clinical Pharmacokinetics and Pharmacodynamics. Clin. Pharmacokinet. 2020, 59, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Parsad, D.; Pandhi, R.; Dogra, S. A guide to selection and appropriate use of macrolides in skin infections. Am. J. Clin. Dermatol. 2003, 4, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Rapp, R.P. Pharmacokinetics and pharmacodynamics of intravenous and oral azithromycin: Enhanced tissue activity and minimal drug interactions. Ann. Pharmacother. 1998, 32, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Pitman, S.K.; Hoang, U.T.P.; Wi, C.H.; Alsheikh, M.; Hiner, D.A.; Percival, K.M. Revisiting Oral Fluoroquinolone and Multivalent Cation Drug-Drug Interactions: Are They Still Relevant? Antibiotics 2019, 8, 108. [Google Scholar] [CrossRef] [Green Version]
- Kays, M.B.; Overholser, B.R.; Mueller, B.A.; Moe, S.M.; Sowinski, K.M. Effects of sevelamer hydrochloride and calcium acetate on the oral bioavailability of ciprofloxacin. Am. J. Kidney Dis. 2003, 42, 1253–1259. [Google Scholar] [CrossRef]
- Bolhuis, M.S.; Panday, P.N.; Pranger, A.D.; Kosterink, J.G.; Alffenaar, J.W. Pharmacokinetic drug interactions of antimicrobial drugs: A systematic review on oxazolidinones, rifamycines, macrolides, fluoroquinolones, and Beta-lactams. Pharmaceutics 2011, 3, 865–913. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Honda, K.; Fujita, K.; Miyachi, Y.; Isoda, K.; Misaka, K.; Suga, Y.; Kato, S.; Tsuchiya, H.; Kato, Y.; et al. Effect of coadministration of rifampicin on the pharmacokinetics of linezolid: Clinical and animal studies. J. Pharm. Health Care Sci. 2018, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Gervasoni, C.; Simonetti, F.R.; Resnati, C.; Charbe, N.; Clementi, E.; Cattaneo, D. Prolonged inductive effect of rifampicin on linezolid exposure. Eur. J. Clin. Pharmacol. 2015, 71, 643–644. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, F.; Tsuji, Y.; Seto, Y.; Ogami, C.; Yamamoto, Y.; To, H. Effects of a rifampicin pre-treatment on linezolid pharmacokinetics. PLoS ONE 2019, 14, e0214037. [Google Scholar] [CrossRef] [PubMed]
- Sisson, T.L.; Jungbluth, G.L.; Hopkins, N.K. A pharmacokinetic evaluation of concomitant administration of linezolid and aztreonam. J. Clin. Pharmacol. 1999, 39, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Cojutti, P.; Maximova, N.; Crichiutti, G.; Isola, M.; Pea, F. Pharmacokinetic/pharmacodynamic evaluation of linezolid in hospitalized paediatric patients: A step toward dose optimization by means of therapeutic drug monitoring and Monte Carlo simulation. J. Antimicrob. Chemother. 2015, 70, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Sivextro-Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/sivextro-epar-product-information_en.pdf (accessed on 11 August 2022).
- Avent, M.L.; Rogers, B.A.; Cheng, A.C.; Paterson, D.L. Current use of aminoglycosides: Indications, pharmacokinetics and monitoring for toxicity. Intern. Med. J. 2011, 41, 441–449. [Google Scholar] [CrossRef]
- Saravolatz, L.D.; Stein, G.E. Plazomicin: A New Aminoglycoside. Clin. Infect. Dis. 2020, 70, 704–709. [Google Scholar] [CrossRef]
- Choi, T.; Komirenko, A.S.; Riddle, V.; Kim, A.; Dhuria, S.V. No Effect of Plazomicin on the Pharmacokinetics of Metformin in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2019, 8, 818–826. [Google Scholar] [CrossRef]
- Libecco, J.A.; Powell, K.R. Trimethoprim/sulfamethoxazole: Clinical update. Pediatr. Rev. 2004, 25, 375–380. [Google Scholar] [CrossRef]
- Regazzi, M.; Carvalho, A.C.; Villani, P.; Matteelli, A. Treatment optimization in patients co-infected with HIV and Mycobacterium tuberculosis infections: Focus on drug-drug interactions with rifamycins. Clin. Pharmacokinet. 2014, 53, 489–507. [Google Scholar] [CrossRef]
- Baciewicz, A.M.; Chrisman, C.R.; Finch, C.K.; Self, T.H. Update on rifampin, rifabutin, and rifapentine drug interactions. Curr. Med. Res. Opin. 2013, 29, 1–12. [Google Scholar] [CrossRef]
- Munsiff, S.S.; Kambili, C.; Ahuja, S.D. Rifapentine for the treatment of pulmonary tuberculosis. Clin. Infect. Dis. 2006, 43, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Keung, A.; Reith, K.; Eller, M.G.; McKenzie, K.A.; Cheng, L.; Weir, S.J. Enzyme induction observed in healthy volunteers after repeated administration of rifapentine and its lack of effect on steady-state rifapentine pharmacokinetics: Part I. Int. J. Tuberc. Lung Dis. 1999, 3, 426–436. [Google Scholar] [PubMed]
- Morte-Romea, E.; Luque-Gómez, P.; Arenere-Mendoza, M.; Sierra-Monzón, J.L.; Pueyo, A.C.; Sagastizabal, G.P.; Muñoz, G.V.; Sánchez Fabra, D.; Paño-Pardo, J.R. Performance Assessment of Software to Detect and Assist Prescribers with Antimicrobial Drug Interactions: Are all of them Created Equal? Antibiotics 2020, 9, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Aziz, M.H.; Alffenaar, J.C.; Bassetti, M.; Bracht, H.; Dimopoulos, G.; Marriott, D.; Neely, M.N.; Paiva, J.A.; Pea, F.; Sjovall, F.; et al. Antimicrobial therapeutic drug monitoring in critically ill adult patients: A Position Paper. Intensive Care Med. 2020, 46, 1127–1153. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.A.; Reddy, M.B.; Heikkinen, A.T.; Lukacova, V.; Parrott, N. Physiologically Based Pharmacokinetic Modelling for First-In-Human Predictions: An Updated Model Building Strategy Illustrated with Challenging Industry Case Studies. Clin. Pharmacokinet. 2019, 58, 727–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asaumi, R.; Nunoya, K.I.; Yamaura, Y.; Taskar, K.S.; Sugiyama, Y. Robust physiologically based pharmacokinetic model of rifampicin for predicting drug-drug interactions via P-glycoprotein induction and inhibition in the intestine, liver, and kidney. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 919–933. [Google Scholar] [CrossRef]
- Yamazaki, S.; Costales, C.; Lazzaro, S.; Eatemadpour, S.; Kimoto, E.; Varma, M.V. Physiologically-Based Pharmacokinetic Modeling Approach to Predict Rifampin-Mediated Intestinal P-Glycoprotein Induction. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Radke, C.; Horn, D.; Lanckohr, C.; Ellger, B.; Meyer, M.; Eissing, T.; Hempel, G. Development of a Physiologically Based Pharmacokinetic Modelling Approach to Predict the Pharmacokinetics of Vancomycin in Critically Ill Septic Patients. Clin. Pharmacokinet. 2017, 56, 759–779. [Google Scholar] [CrossRef]
- Takita, H.; Barnett, S.; Zhang, Y.; Ménochet, K.; Shen, H.; Ogungbenro, K.; Galetin, A. PBPK Model of Coproporphyrin I: Evaluation of the Impact of SLCO1B1 Genotype, Ethnicity, and Sex on its Inter-Individual Variability. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 137–147. [Google Scholar] [CrossRef]
- Jann, M.W.; Cohen, L.J. The influence of ethnicity and antidepressant pharmacogenetics in the treatment of depression. Drug Metabol. Drug Interact. 2000, 16, 39–67. [Google Scholar] [CrossRef]
- Fitzmaurice, M.G.; Wong, A.; Akerberg, H.; Avramovska, S.; Smithburger, P.L.; Buckley, M.S.; Kane-Gill, S.L. Evaluation of Potential Drug-Drug Interactions in Adults in the Intensive Care Unit: A Systematic Review and Meta-Analysis. Drug. Saf. 2019, 42, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Aghili, M.; Kasturirangan, M.N. Management of Drug-Drug Interactions among Critically Ill Patients with Chronic Kidney Disease: Impact of Clinical Pharmacist’s Interventions. Indian J. Crit. Care Med. 2021, 25, 1226–1231. [Google Scholar] [CrossRef] [PubMed]

| Antibiotic | Inhibitory/Inducing Effects | Victim of pDDIs | Perpetrator of pDDIs | Comments |
|---|---|---|---|---|
| Ceftobiprol | Inhibitor of OATP1B1 and OATP1B3 | None | May increase the disposition of OATP substrates | Clinical relevance not demonstrated |
| Ceftaroline | None | None | None | None |
| Cefiderocol | Inhibitor of OAT1, OAT3, OCT1, OCT2, MATE-2K, OATP1B3 | None | May increase the disposition of substrates of drug transporters | Clinical relevance not demonstrated |
| Ceftolozane | None | None | None | None |
| Dalbavancin | None | None | None | The effects of inhibitors on drug transporters have not been studied |
| Oritavancin | Weak inhibitor of CYP2C9 and CYP2C19, inducer of CYP3A4 and CYP2D6 | None | May increase CYP2C9/2C19 substrates and reduce CYP3A4/2D6 substrates | Administer with caution with NTI drugs metabolized by these enzymes |
| Telavancin | None | None | None | The effects of inhibitors on drug transporters have not been studied |
| Plazomicin | Inhibitor of MATE2-K, MATE1, OCT2 | None | May increase the disposition of substrates of drug transporters | Clinical relevance not demonstrated |
| Eravacycline | None | Drug exposure reduced by strong CYP3A inducers | None | Increase drug dose of (i.e., 1.5 mg/kg bid) when given with a strong CYP3A inducer |
| Tedizolid | Inhibition of BCRP | None | May increase the disposition of BCRP substrates | Clinical relevance not demonstrated |
| Link | Notes |
|---|---|
| https://clinicalweb.marionegri.it/intercheckweb | A database that evaluates prescriptive appropriateness in the elderly by considering various aspects of geriatric pharmacology |
| https://reference.medscape.com/drug-interactionchecker | A “generalist” database that also includes over-the-counter products, some phytotherapeutic agents and supplements |
| https://www.hiv-druginteractions.org | A database verifying interactions between antiretroviral agents (HIV), and between antiretroviral and non-antiretroviral agents |
| https://www.hep-druginteractions.org | A database verifying interactions between antiviral agents (HCV), and between antiviral and non-antiviral agents |
| http://www.drugs.com/drug_interactions.html | A “generalist” database |
| https://cancer-druginteractions.org/checker | A database verifying interactions between antitumor agents, and between antitumor and non-antitumor agents |
| http://healthlibrary.uchospitals.edu/Library/DrugReference/DrugInteraction/ | A “generalist” database |
| https://www.rxlist.com/drug-interaction-checker.htm | A “generalist” database |
| https://www.ddi-predictor.org/predictor/ddi | A “generalist” database |
| https://stahlonline.cambridge.org/drug_interaction.jsf?page=drugDetails | A “generalist” database that particularly focuses on drugs acting on the central nervous system |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cattaneo, D.; Gervasoni, C.; Corona, A. The Issue of Pharmacokinetic-Driven Drug-Drug Interactions of Antibiotics: A Narrative Review. Antibiotics 2022, 11, 1410. https://doi.org/10.3390/antibiotics11101410
Cattaneo D, Gervasoni C, Corona A. The Issue of Pharmacokinetic-Driven Drug-Drug Interactions of Antibiotics: A Narrative Review. Antibiotics. 2022; 11(10):1410. https://doi.org/10.3390/antibiotics11101410
Chicago/Turabian StyleCattaneo, Dario, Cristina Gervasoni, and Alberto Corona. 2022. "The Issue of Pharmacokinetic-Driven Drug-Drug Interactions of Antibiotics: A Narrative Review" Antibiotics 11, no. 10: 1410. https://doi.org/10.3390/antibiotics11101410