
Long-Read Whole Genome Sequencing Elucidates the Mechanisms of Amikacin Resistance in Multidrug-Resistant Klebsiella pneumoniae Isolates Obtained from COVID-19 Patients
 , ,
, ,
Abstract
:1. Introduction
2. Results
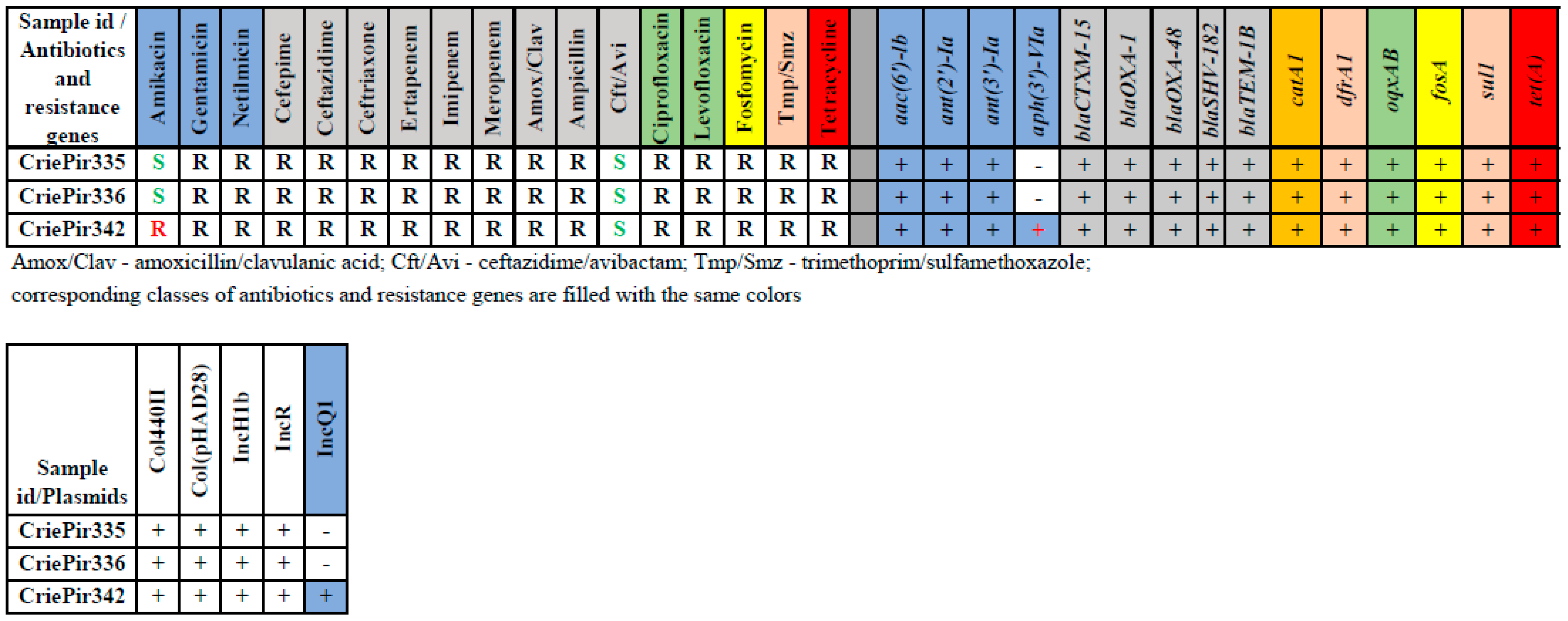
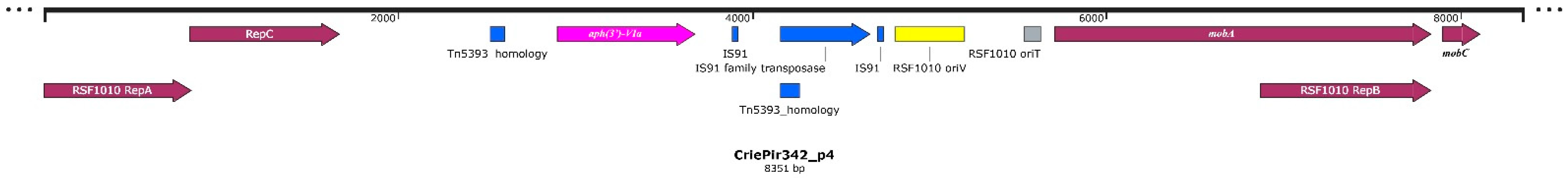
2.1. Isolate Typing, Resistance Profile, AMR Gene and Plasmid Content Determination
2.2. Virulence Factors
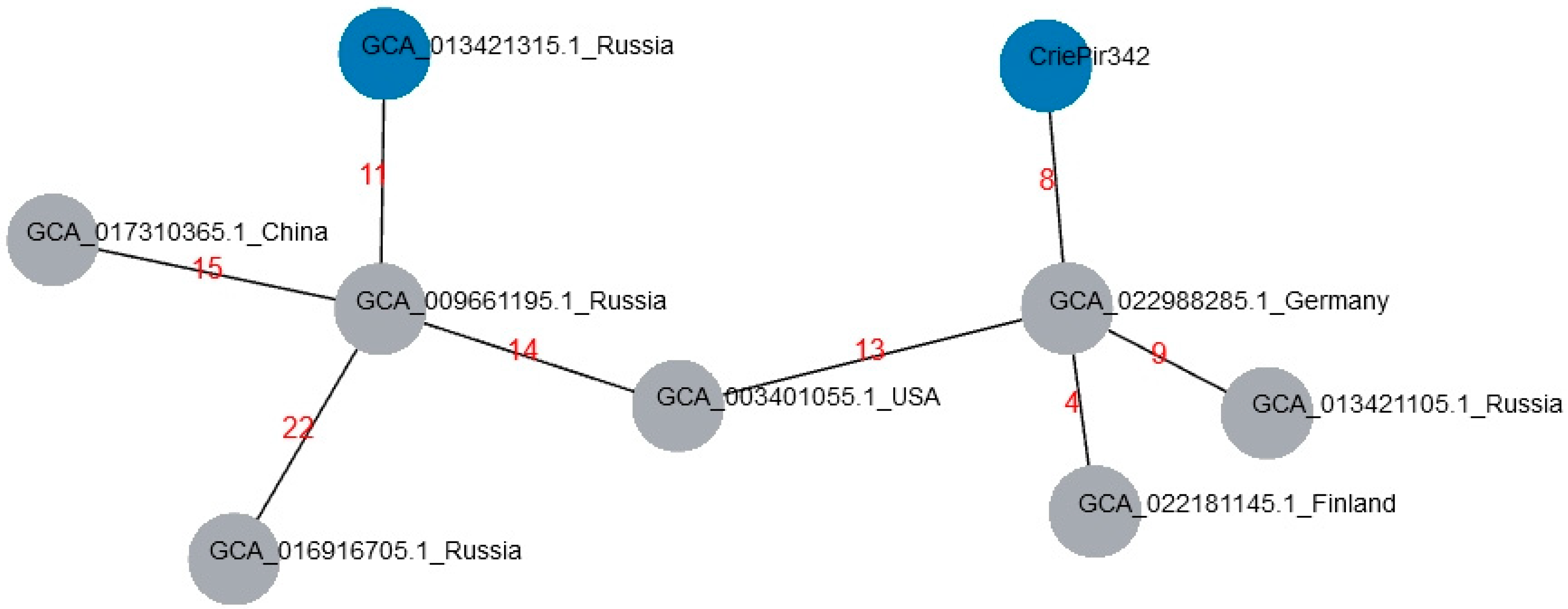
2.3. Comparison with Reference Isolates of ST395 from Genbank Database
3. Discussion
4. Materials and Methods
4.1. Sample Collection, Susceptibility Testing, DNA Isolation, and Sequencing
4.2. Data Processing and Genome Assembly
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, M.; Earley, M.; Chen, L.; Hanson, B.M.; Yu, Y.; Liu, Z.; Salcedo, S.; Cober, E.; Li, L.; Kanj, S.S.; et al. Clinical outcomes and bacterial characteristics of carbapenem-resistant Klebsiella pneumoniae complex among patients from different global regions (CRACKLE-2): A prospective, multicentre, cohort study. Lancet Infect. Dis. 2022, 22, 401–412. [Google Scholar] [CrossRef]
- Manesh, A.; Varghese, G.M. Rising antimicrobial resistance: An evolving epidemic in a pandemic. Lancet Microbe 2021, 2, e419–e420. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Agyeman, A.A.; Bergen, P.J.; Rao, G.G.; Nation, R.L.; Landersdorfer, C.B. A systematic review and meta-analysis of treatment outcomes following antibiotic therapy among patients with carbapenem-resistant Klebsiella pneumoniae infections. Int. J. Antimicrob. Agents 2020, 55, 105833. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Hu, T.; Hu, J.; Song, N.; Zhang, Y.; Chen, Y. Five-year change of prevalence and risk factors for infection and mortality of carbapenem-resistant Klebsiella pneumoniae bloodstream infection in a tertiary hospital in North China. Antimicrob. Resist. Infect. Control 2020, 9, 79. [Google Scholar] [CrossRef]
- Gorrie, C.L.; Mirceta, M.; Wick, R.R.; Judd, L.M.; Lam, M.M.C.; Gomi, R.; Abbott, I.J.; Thomson, N.R.; Strugnell, R.A.; Pratt, N.F.; et al. Genomic dissection of Klebsiella pneumoniae infections in hospital patients reveals insights into an opportunistic pathogen. Nat. Commun. 2022, 13, 3017. [Google Scholar] [CrossRef]
- Saini, V.; Jain, C.; Singh, N.P.; Alsulimani, A.; Gupta, C.; Dar, S.A.; Haque, S.; Das, S. Paradigm Shift in Antimicrobial Resistance Pattern of Bacterial Isolates during the COVID-19 Pandemic. Antibiotics 2021, 10, 954. [Google Scholar] [CrossRef]
- Lai, C.C.; Chen, S.Y.; Ko, W.C.; Hsueh, P.R. Increased antimicrobial resistance during the COVID-19 pandemic. Int. J. Antimicrob. Agents 2021, 57, 106324. [Google Scholar] [CrossRef]
- Pitout, J.D.; Nordmann, P.; Poirel, L. Carbapenemase-Producing Klebsiella pneumoniae, a Key Pathogen Set for Global Nosocomial Dominance. Antimicrob. Agents Chemother. 2015, 59, 5873–5884. [Google Scholar] [CrossRef] [Green Version]
- Spaziante, M.; Oliva, A.; Ceccarelli, G.; Venditti, M. What are the treatment options for resistant Klebsiella pneumoniae carbapenemase (KPC)-producing bacteria? Expert Opin. Pharmacother. 2020, 21, 1781–1787. [Google Scholar] [CrossRef]
- Tsuji, B.T.; Pogue, J.M.; Zavascki, A.P.; Paul, M.; Daikos, G.L.; Forrest, A.; Giacobbe, D.R.; Viscoli, C.; Giamarellou, H.; Karaiskos, I.; et al. International Consensus Guidelines for the Optimal Use of the Polymyxins: Endorsed by the American College of Clinical Pharmacy (ACCP), European Society of Clinical Microbiology and Infectious Diseases (ESCMID), Infectious Diseases Society of America (IDSA), International Society for Anti-infective Pharmacology (ISAP), Society of Critical Care Medicine (SCCM), and Society of Infectious Diseases Pharmacists (SIDP). Pharmacotherapy 2019, 39, 10–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhao, J.; Ji, F.; Chang, H.; Qin, J.; Zhang, C.; Hu, G.; Zhu, J.; Yang, J.; Jia, Z.; et al. Multiple-Replicon Resistance Plasmids of Klebsiella Mediate Extensive Dissemination of Antimicrobial Genes. Front. Microbiol. 2021, 12, 754931. [Google Scholar] [CrossRef]
- Meng, M.; Li, Y.; Yao, H. Plasmid-Mediated Transfer of Antibiotic Resistance Genes in Soil. Antibiotics 2022, 11, 525. [Google Scholar] [CrossRef] [PubMed]
- Shaidullina, E.; Shelenkov, A.; Yanushevich, Y.; Mikhaylova, Y.; Shagin, D.; Alexandrova, I.; Ershova, O.; Akimkin, V.; Kozlov, R.; Edelstein, M. Antimicrobial Resistance and Genomic Characterization of OXA-48- and CTX-M-15-Co-Producing Hypervirulent Klebsiella pneumoniae ST23 Recovered from Nosocomial Outbreak. Antibiotics 2020, 9, 862. [Google Scholar] [CrossRef]
- Peter, S.; Bosio, M.; Gross, C.; Bezdan, D.; Gutierrez, J.; Oberhettinger, P.; Liese, J.; Vogel, W.; Dorfel, D.; Berger, L.; et al. Tracking of Antibiotic Resistance Transfer and Rapid Plasmid Evolution in a Hospital Setting by Nanopore Sequencing. mSphere 2020, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Bird, M.T.; Greig, D.R.; Nair, S.; Jenkins, C.; Godbole, G.; Gharbia, S.E. Use of Nanopore Sequencing to Characterise the Genomic Architecture of Mobile Genetic Elements Encoding blaCTX-M-15 in Escherichia coli Causing Travellers’Diarrhoea. Front. Microbiol. 2022, 13, 862234. [Google Scholar] [CrossRef]
- Ferreira, F.A.; Helmersen, K.; Visnovska, T.; Jorgensen, S.B.; Aamot, H.V. Rapid nanopore-based DNA sequencing protocol of antibiotic-resistant bacteria for use in surveillance and outbreak investigation. Microb. Genom. 2021, 7, 000557. [Google Scholar] [CrossRef] [PubMed]
- Schurch, A.C.; Arredondo-Alonso, S.; Willems, R.J.L.; Goering, R.V. Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene-based approaches. Clin. Microbiol. Infect. 2018, 24, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Lambert, T.; Gerbaud, G.; Courvalin, P. Characterization of transposon Tn1528, which confers amikacin resistance by synthesis of aminoglycoside 3’-O-phosphotransferase type VI. Antimicrob. Agents Chemother. 1994, 38, 702–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzmenkov, A.Y.; Trushin, I.V.; Vinogradova, A.G.; Avramenko, A.A.; Sukhorukova, M.V.; Malhotra-Kumar, S.; Dekhnich, A.V.; Edelstein, M.V.; Kozlov, R.S. AMRmap: An Interactive Web Platform for Analysis of Antimicrobial Resistance Surveillance Data in Russia. Front. Microbiol. 2021, 12, 620002. [Google Scholar] [CrossRef]
- Dogan, O.; Vatansever, C.; Atac, N.; Albayrak, O.; Karahuseyinoglu, S.; Sahin, O.E.; Kilicoglu, B.K.; Demiray, A.; Ergonul, O.; Gonen, M.; et al. Virulence Determinants of Colistin-Resistant K. pneumoniae High-Risk Clones. Biology 2021, 10, 436. [Google Scholar] [CrossRef]
- Muggeo, A.; Guillard, T.; Klein, F.; Reffuveille, F.; Francois, C.; Babosan, A.; Bajolet, O.; Bertrand, X.; de Champs, C.; CarbaFrEst, G. Spread of Klebsiella pneumoniae ST395 non-susceptible to carbapenems and resistant to fluoroquinolones in North-Eastern France. J. Glob. Antimicrob. Resist. 2018, 13, 98–103. [Google Scholar] [CrossRef]
- Kovacs, K.; Nyul, A.; Mestyan, G.; Melegh, S.; Fenyvesi, H.; Jakab, G.; Szabo, H.; Janvari, L.; Damjanova, I.; Toth, A. Emergence and interhospital spread of OXA-48-producing Klebsiella pneumoniae ST395 clone in Western Hungary. Infect. Dis. 2017, 49, 231–233. [Google Scholar] [CrossRef]
- Cienfuegos-Gallet, A.V.; Zhou, Y.; Ai, W.; Kreiswirth, B.N.; Yu, F.; Chen, L. Multicenter Genomic Analysis of Carbapenem-Resistant Klebsiella pneumoniae from Bacteremia in China. Microbiol. Spectr. 2022, 10, e0229021. [Google Scholar] [CrossRef] [PubMed]
- Shelenkov, A.; Mikhaylova, Y.; Yanushevich, Y.; Samoilov, A.; Petrova, L.; Fomina, V.; Gusarov, V.; Zamyatin, M.; Shagin, D.; Akimkin, V. Molecular Typing, Characterization of Antimicrobial Resistance, Virulence Profiling and Analysis of Whole-Genome Sequence of Clinical Klebsiella pneumoniae Isolates. Antibiotics 2020, 9, 261. [Google Scholar] [CrossRef] [PubMed]
- Muraya, A.; Kyany’a, C.; Kiyaga, S.; Smith, H.J.; Kibet, C.; Martin, M.J.; Kimani, J.; Musila, L. Antimicrobial Resistance and Virulence Characteristics of Klebsiella pneumoniae Isolates in Kenya by Whole-Genome Sequencing. Pathogens 2022, 11, 545. [Google Scholar] [CrossRef] [PubMed]
- Lery, L.M.; Frangeul, L.; Tomas, A.; Passet, V.; Almeida, A.S.; Bialek-Davenet, S.; Barbe, V.; Bengoechea, J.A.; Sansonetti, P.; Brisse, S.; et al. Comparative analysis of Klebsiella pneumoniae genomes identifies a phospholipase D family protein as a novel virulence factor. BMC Biol. 2014, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, T.; Chen, L.; Du, H. Virulence Factors in Hypervirulent Klebsiella pneumoniae. Front. Microbiol. 2021, 12, 642484. [Google Scholar] [CrossRef] [PubMed]
- Arbatsky, N.P.; Shneider, M.M.; Dmitrenok, A.S.; Popova, A.V.; Shagin, D.A.; Shelenkov, A.A.; Mikhailova, Y.V.; Edelstein, M.V.; Knirel, Y.A. Structure and gene cluster of the K125 capsular polysaccharide from Acinetobacter baumannii MAR13-1452. Int. J. Biol. Macromol. 2018, 117, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Heinz, E.; Holt, K.E.; Wyres, K.L. Kaptive Web: User-Friendly Capsule and Lipopolysaccharide Serotype Prediction for Klebsiella Genomes. J. Clin. Microbiol. 2018, 56, e00197-18. [Google Scholar] [CrossRef]
- Arena, F.; Menchinelli, G.; Di Pilato, V.; Torelli, R.; Antonelli, A.; Henrici De Angelis, L.; Coppi, M.; Sanguinetti, M.; Rossolini, G.M. Resistance and virulence features of hypermucoviscous Klebsiella pneumoniae from bloodstream infections: Results of a nationwide Italian surveillance study. Front. Microbiol. 2022, 13, 983294. [Google Scholar] [CrossRef]
- Alekseeva, A.E.; Brusnigina, N.F.; Gordinskaya, N.A.; Makhova, M.A.; Kolesnikova, E.A. Molecular genetic characteristics of resistome and virulome of carbapenem-resistant Klebsiella pneumoniae clinical strains. Klin. Lab. Diagn. 2022, 67, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef] [Green Version]
- Orlek, A.; Phan, H.; Sheppard, A.E.; Doumith, M.; Ellington, M.; Peto, T.; Crook, D.; Walker, A.S.; Woodford, N.; Anjum, M.F.; et al. Ordering the mob: Insights into replicon and MOB typing schemes from analysis of a curated dataset of publicly available plasmids. Plasmid 2017, 91, 42–52. [Google Scholar] [CrossRef]
- Meyer, R. Replication and conjugative mobilization of broad host-range IncQ plasmids. Plasmid 2009, 62, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loftie-Eaton, W.; Rawlings, D.E. Diversity, biology and evolution of IncQ-family plasmids. Plasmid 2012, 67, 15–34. [Google Scholar] [CrossRef]
- Prussing, C.; Snavely, E.A.; Singh, N.; Lapierre, P.; Lasek-Nesselquist, E.; Mitchell, K.; Haas, W.; Owsiak, R.; Nazarian, E.; Musser, K.A. Nanopore MinION Sequencing Reveals Possible Transfer of bla KPC-2 Plasmid Across Bacterial Species in Two Healthcare Facilities. Front. Microbiol. 2020, 11, 2007. [Google Scholar] [CrossRef] [PubMed]
- Greig, D.R.; Jenkins, C.; Gharbia, S.E.; Dallman, T.J. Analysis of a small outbreak of Shiga toxin-producing Escherichia coli O157:H7 using long-read sequencing. Microb. Genom. 2021, 7, 000545. [Google Scholar] [CrossRef]
- Roberts, L.W.; Harris, P.N.A.; Forde, B.M.; Ben Zakour, N.L.; Catchpoole, E.; Stanton-Cook, M.; Phan, M.D.; Sidjabat, H.E.; Bergh, H.; Heney, C.; et al. Integrating multiple genomic technologies to investigate an outbreak of carbapenemase-producing Enterobacter hormaechei. Nat. Commun. 2020, 11, 466. [Google Scholar] [CrossRef] [Green Version]
- Steinig, E.; Duchene, S.; Aglua, I.; Greenhill, A.; Ford, R.; Yoannes, M.; Jaworski, J.; Drekore, J.; Urakoko, B.; Poka, H.; et al. Phylodynamic Inference of Bacterial Outbreak Parameters Using Nanopore Sequencing. Mol. Biol. Evol. 2022, 39, 3. [Google Scholar] [CrossRef]
- de Siqueira, G.M.V.; Pereira-Dos-Santos, F.M.; Silva-Rocha, R.; Guazzaroni, M.E. Nanopore Sequencing Provides Rapid and Reliable Insight Into Microbial Profiles of Intensive Care Units. Front. Public Health 2021, 9, 710985. [Google Scholar] [CrossRef]
- Shelenkov, A.; Petrova, L.; Zamyatin, M.; Mikhaylova, Y.; Akimkin, V. Diversity of International High-Risk Clones of Acinetobacter baumannii Revealed in a Russian Multidisciplinary Medical Center during 2017–2019. Antibiotics 2021, 10, 1009. [Google Scholar] [CrossRef] [PubMed]
- Mustapha, M.M.; Srinivasa, V.R.; Griffith, M.P.; Cho, S.T.; Evans, D.R.; Waggle, K.; Ezeonwuka, C.; Snyder, D.J.; Marsh, J.W.; Harrison, L.H.; et al. Genomic Diversity of Hospital-Acquired Infections Revealed through Prospective Whole-Genome Sequencing-Based Surveillance. mSystems 2022, 7, e0138421. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; De Maio, N.; Lanfear, R.; MacCannell, D.R.; Minh, B.Q.; Schmidt, H.A.; Stamatakis, A.; Goldman, N.; Dessimoz, C. Want to track pandemic variants faster? Fix the bioinformatics bottleneck. Nature 2021, 591, 30–33. [Google Scholar] [CrossRef]
- Viehweger, A.; Blumenscheit, C.; Lippmann, N.; Wyres, K.L.; Brandt, C.; Hans, J.B.; Holzer, M.; Irber, L.; Gatermann, S.; Lubbert, C.; et al. Context-aware genomic surveillance reveals hidden transmission of a carbapenemase-producing Klebsiella pneumoniae. Microb. Genom. 2021, 7, 000741. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoSComput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feijao, P.; Yao, H.T.; Fornika, D.; Gardy, J.; Hsiao, W.; Chauve, C.; Chindelevitch, L. MentaLiST-A fast MLST caller for large MLST schemes. Microb. Genom. 2018, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Tang, H. ISEScan: Automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Id | Length | RepliconType | RelaxaseFamily | Plasmid Type |
|---|---|---|---|---|
| 2 | 282,773 | IncH | MOBH, MPFT | Conjugative |
| 3 | 74,680 | IncR | - | Non-mobilizable |
| 4 | 8351 | IncQ | MOBQ | Mobilizable |
| 5 | 5010 | ColRNAI | - | Non-mobilizable |
| 6 | 4052 | ColRNAI | MOBP | Mobilizable |
| 7 | 3511 | ColRNAI | MOBP | Mobilizable |
| Gene Cluster | Function | Location |
|---|---|---|
| acrAB | efflux pump genes | Chromosome |
| fimABCDEFGHIK | fimbria production and biofilm formation | Chromosome |
| entABCEF, fepABCDG | enterobactin biosynthesis (siderophore) | Chromosome |
| irp1,2 | iron acquisition system | Chromosome |
| iucABCD, iutA | aerobactin cluster-iron acquisition system | IncH1B plasmid |
| manBC | promoters of capsule synthesis genes | Chromosome |
| mrkABCDFHIJ | fimbria production and biofilm formation | Chromosome |
| rcsAB | exopolysaccharide biosynthesis | Chromosome |
| rmpA2 | regulator of mucoid phenotype | IncH1B plasmid |
| wbbMNO | lipopolysaccharide synthesis | Chromosome |
| ybtAEPQSTU | yersiniabactin cluster-iron acquisition system | Chromosome |
| Genbank Acc. | Number of Allele Differences | Country of Isolation | Isolate Collection Year |
|---|---|---|---|
| GCA_022988285.1 | 8 | Germany | 2016 |
| GCA_022181145.1 | 10 | Finland | 2018 |
| GCA_003401055.1 | 13 | USA | 2013 |
| GCA_013421105.1 | 15 | Russia | 2018 |
| GCA_009661195.1 | 17 | Russia | 2018 |
| GCA_013421315.1 | 18 | Russia | 2018 |
| GCA_017310365.1 | 18 | China | 2015 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shelenkov, A.; Petrova, L.; Mironova, A.; Zamyatin, M.; Akimkin, V.; Mikhaylova, Y. Long-Read Whole Genome Sequencing Elucidates the Mechanisms of Amikacin Resistance in Multidrug-Resistant Klebsiella pneumoniae Isolates Obtained from COVID-19 Patients. Antibiotics 2022, 11, 1364. https://doi.org/10.3390/antibiotics11101364
Shelenkov A, Petrova L, Mironova A, Zamyatin M, Akimkin V, Mikhaylova Y. Long-Read Whole Genome Sequencing Elucidates the Mechanisms of Amikacin Resistance in Multidrug-Resistant Klebsiella pneumoniae Isolates Obtained from COVID-19 Patients. Antibiotics. 2022; 11(10):1364. https://doi.org/10.3390/antibiotics11101364
Chicago/Turabian StyleShelenkov, Andrey, Lyudmila Petrova, Anna Mironova, Mikhail Zamyatin, Vasiliy Akimkin, and Yulia Mikhaylova. 2022. "Long-Read Whole Genome Sequencing Elucidates the Mechanisms of Amikacin Resistance in Multidrug-Resistant Klebsiella pneumoniae Isolates Obtained from COVID-19 Patients" Antibiotics 11, no. 10: 1364. https://doi.org/10.3390/antibiotics11101364