
Evaluation of Heterocyclic Carboxamides as Potential Efflux Pump Inhibitors in Pseudomonas aeruginosa

Abstract
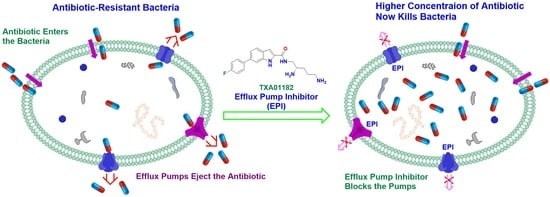
:
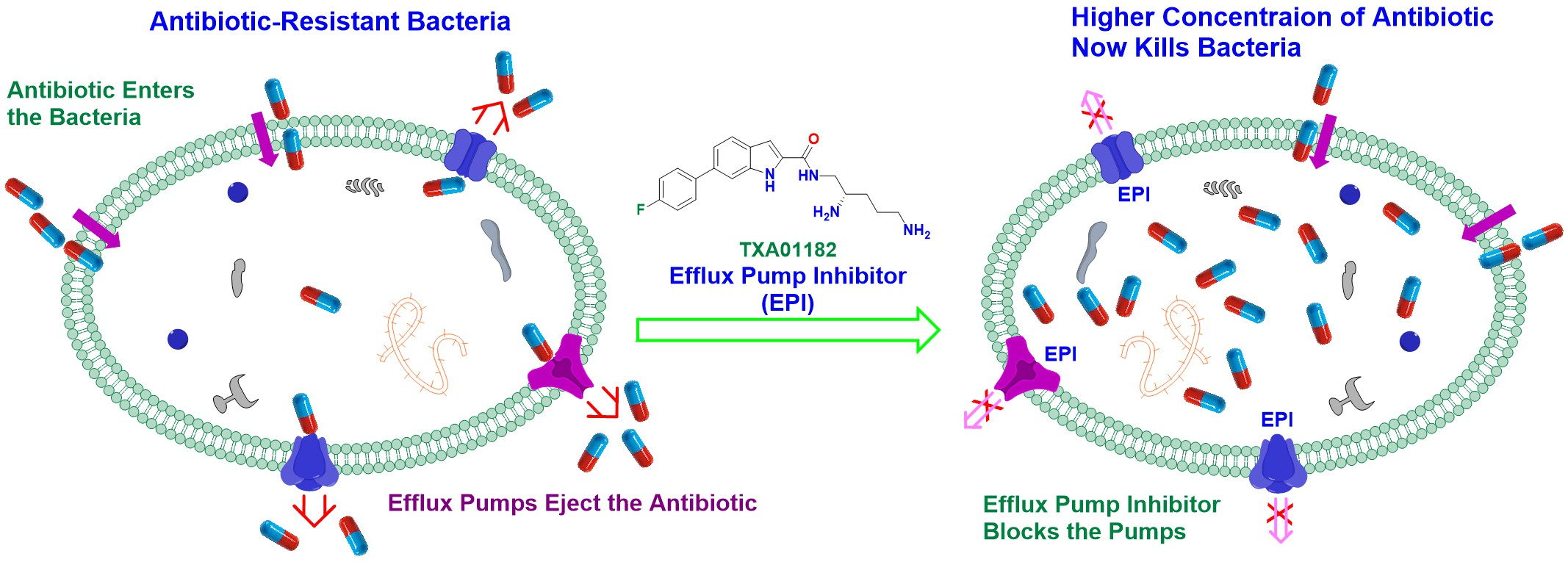
1. Introduction
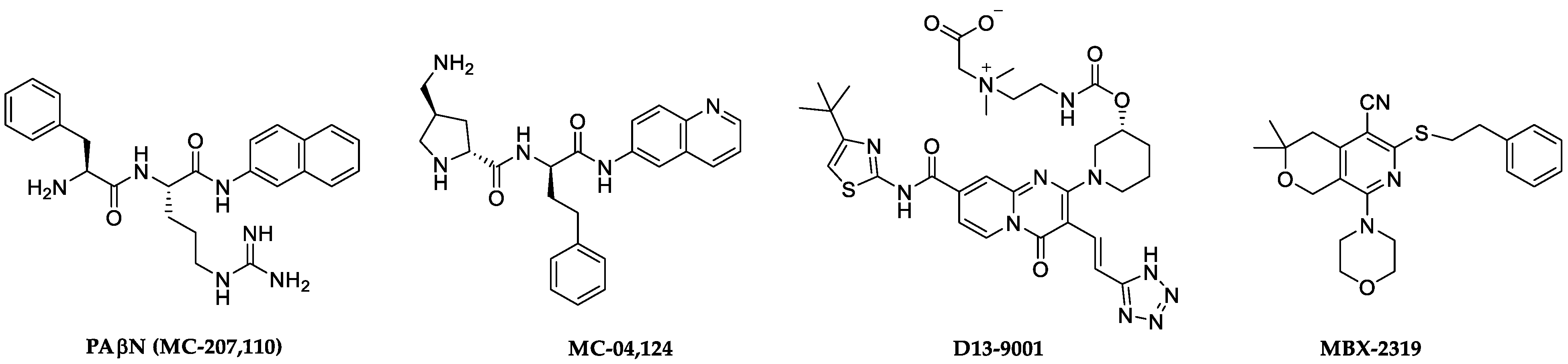
2. Results and Discussion
2.1. Screening Results
2.2. In-Depth Potentiation Evaluation of 6j (TXA01182)
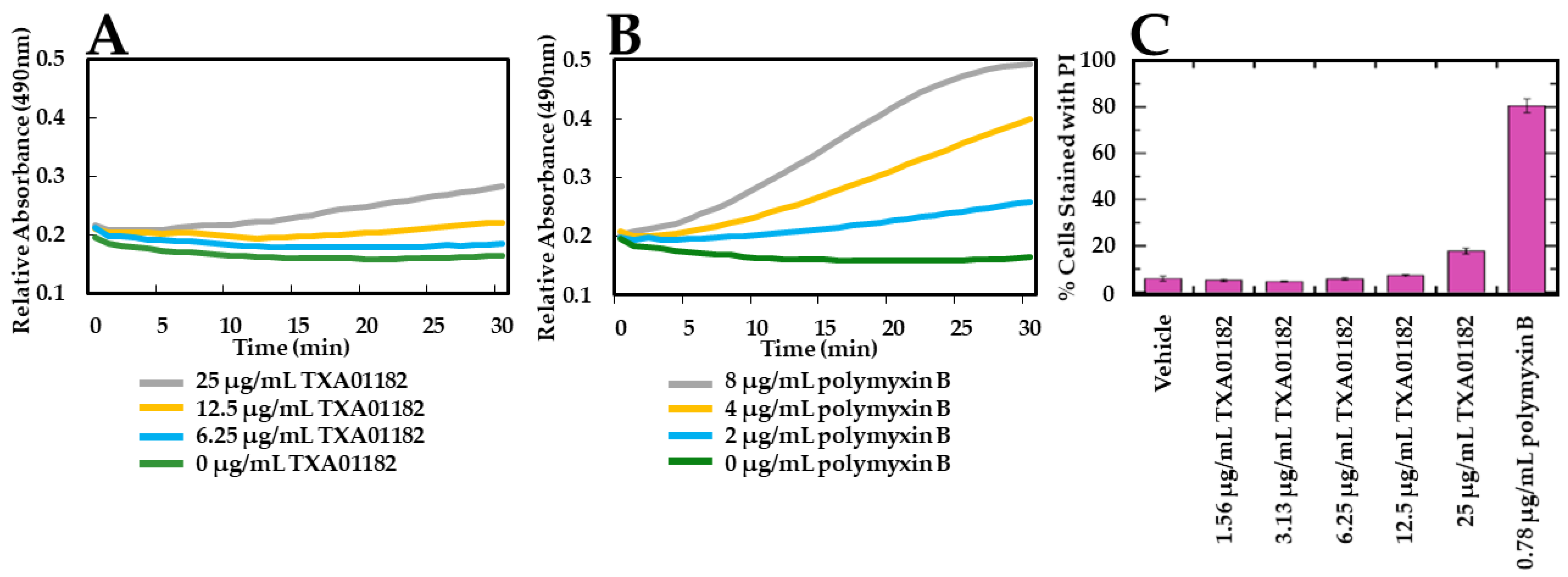
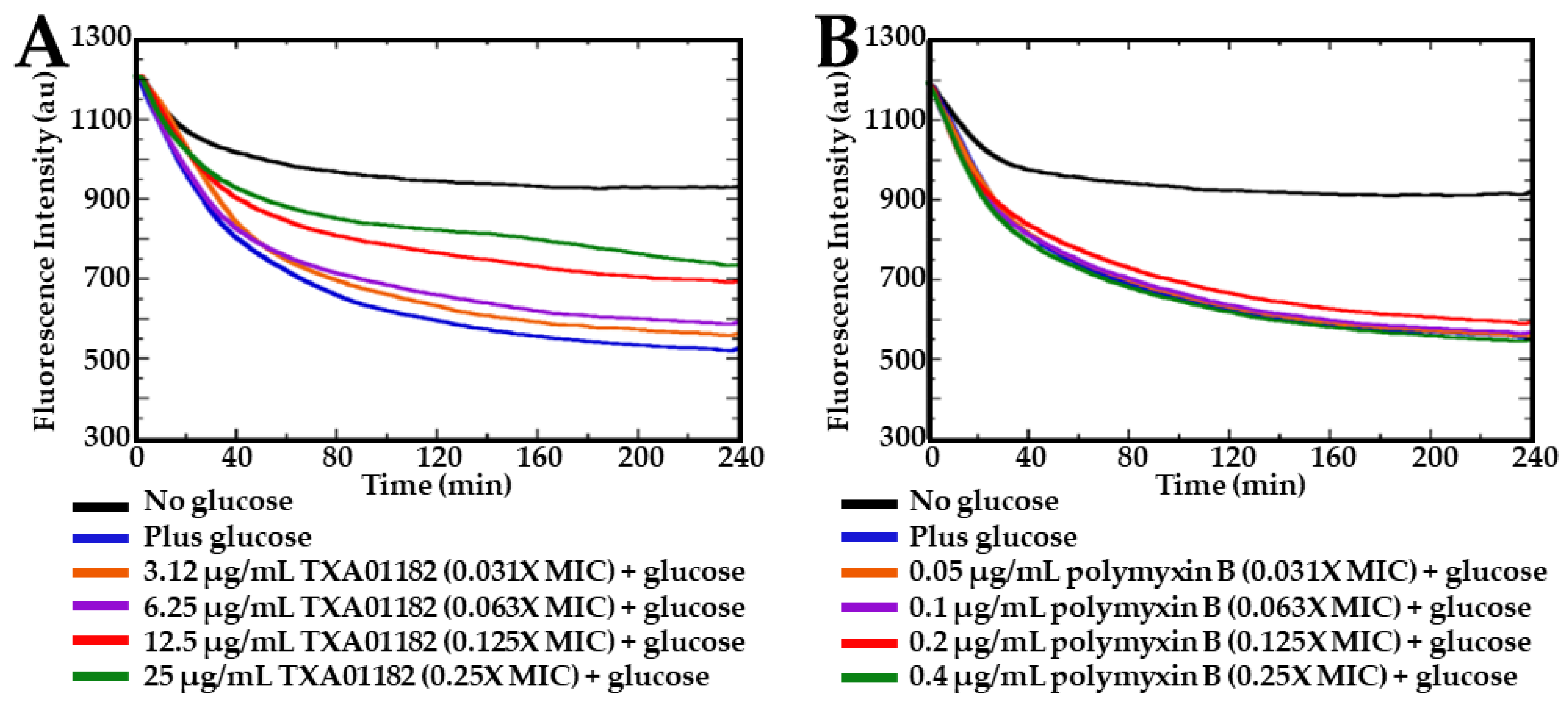
2.3. TXA01182 Plays a Minimal Role in Membrane Disruption
2.4. TXA01182 Inhibits the Efflux of Ethidium Bromide
2.5. TXA01182 Enhances the Activity of Levofloxacin against Clinical Isolates of P. aeruginosa
2.6. TXA01182 Lowers the Frequency of Resistance to Levofloxacin
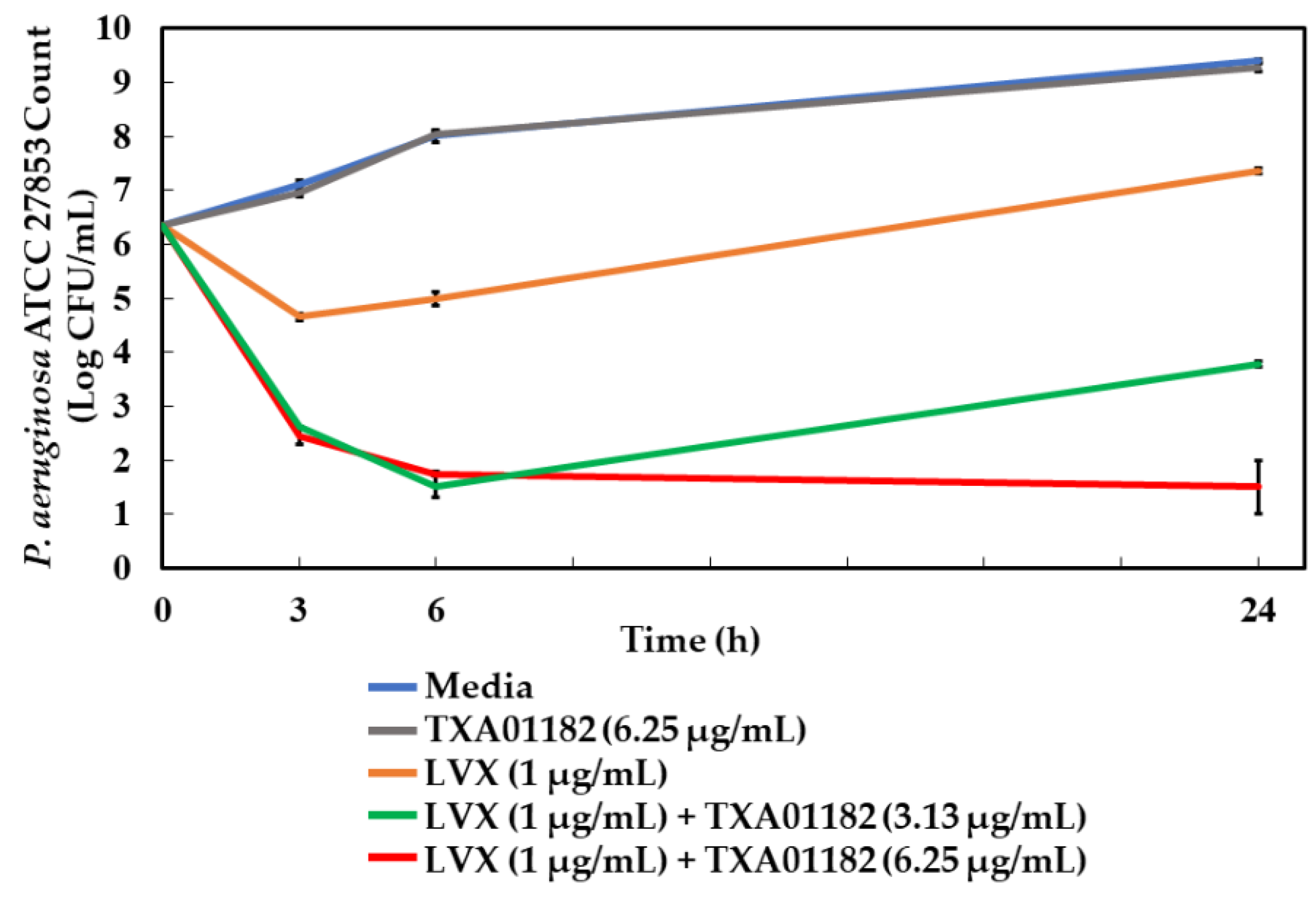
2.7. Time-Kill Assay
3. Materials and Methods
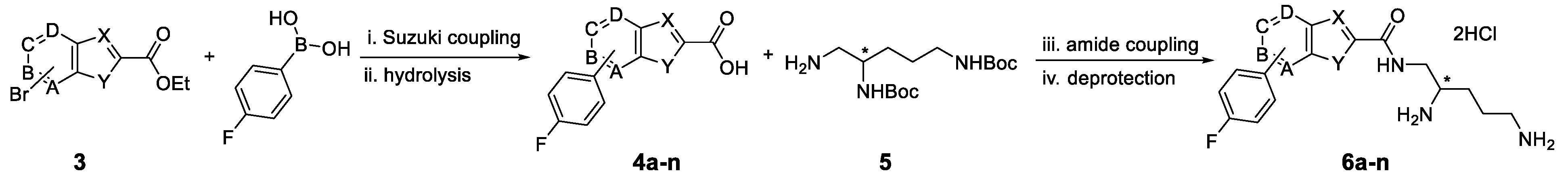
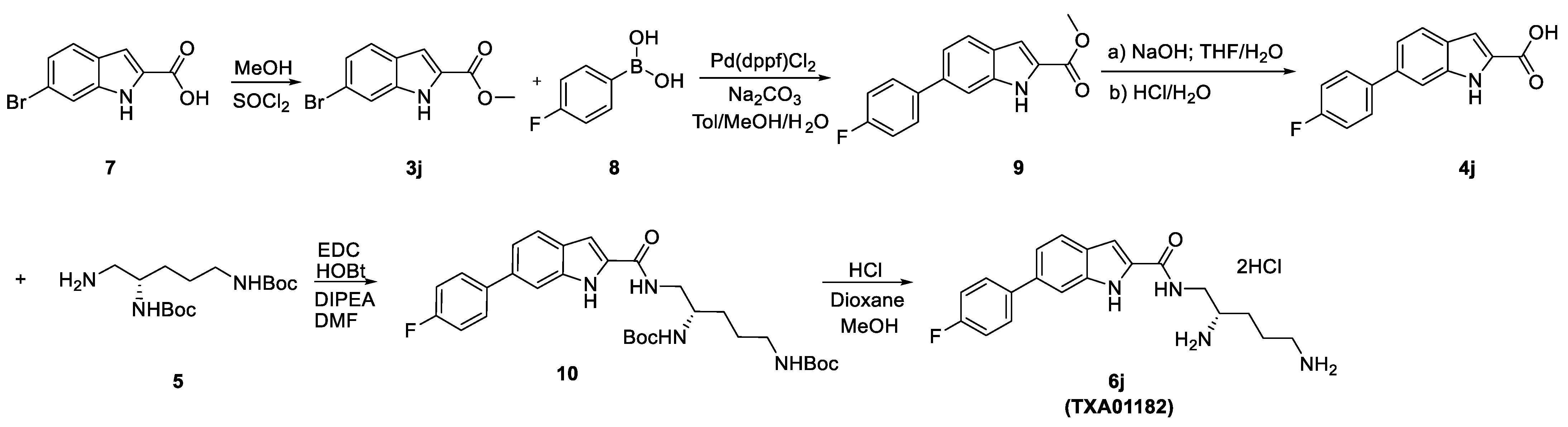
3.1. Synthesis
3.2. Bacterial Strains, Media, and Reagents
3.3. Minimum Inhibitory Concentration (MIC) Assay for Potentiation of Antimicrobial Activity against P. aeruginosa
3.4. Fluorescence-Activated Cell Sorting (FACS) Assay for Permeabilization of the Outer and Inner Cell Membranes to Propidium Iodide (PI) in P. aeruginosa
3.5. Nitrocefin (NCF) Cellular Assay for Outer Cell Membrane Permeabilization Assessment in P. aeruginosa
3.6. Fluorescence-Based Cellular Assay for Inhibition of Pump-Mediated Efflux of Ethidium Bromide (EtBr)
3.7. Frequency of Resistance (FoR) Studies
3.8. Time-Kill Studies
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amaral, L.; Martins, A.; Spengler, G.; Molnar, J. Efflux pumps of Gram-negative bacteria: What they do, how they do it, with what and how to deal with them. Front. Pharmacol. 2014, 4, 168. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria. Drugs 2004, 64, 159–204. [Google Scholar] [CrossRef]
- Nikaido, H.; Pages, J.M. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol. Rev. 2012, 36, 340–363. [Google Scholar] [CrossRef] [Green Version]
- Colclough, A.L.; Alav, I.; Whittle, E.E.; Pugh, H.L.; Darby, E.M.; Legood, S.W.; McNeil, H.E.; Blair, J.M. RND efflux pumps in Gram-negative bacteria; regulation, structure and role in antibiotic resistance. Future Microbiol. 2020, 15, 143–157. [Google Scholar] [CrossRef]
- Daury, L.; Orange, F.; Taveau, J.C.; Verchere, A.; Monlezun, L.; Gounou, C.; Marreddy, R.K.; Picard, M.; Broutin, I.; Pos, K.M.; et al. Tripartite assembly of RND multidrug efflux pumps. Nat. Commun. 2016, 7, 10731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, K. Multidrug efflux pumps and antimicrobial resistance in Pseudomonas aeruginosa and related organisms. J. Mol. Microbiol. Biotechnol. 2001, 3, 255–264. [Google Scholar]
- Poole, K.; Srikumar, R. Multidrug efflux in Pseudomonas aeruginosa: Components, mechanisms and clinical significance. Curr. Top. Med. Chem. 2001, 1, 59–71. [Google Scholar] [CrossRef]
- Serra, C.; Bouharkat, B.; Tir Touil-Meddah, A.; Guenin, S.; Mullie, C. MexXY Multidrug Efflux System Is More Frequently Overexpressed in Ciprofloxacin Resistant French Clinical Isolates Compared to Hospital Environment Ones. Front. Microbiol. 2019, 10, 366. [Google Scholar] [CrossRef]
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-resistant Pseudomonas aeruginosa: Clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef] [Green Version]
- Blanco, P.; Sanz-Garcia, F.; Hernando-Amado, S.; Martinez, J.L.; Alcalde-Rico, M. The development of efflux pump inhibitors to treat Gram-negative infections. Expert Opin. Drug Discov. 2018, 13, 919–931. [Google Scholar] [CrossRef]
- Wang, Y.; Venter, H.; Ma, S. Efflux Pump Inhibitors: A Novel Approach to Combat Efflux-Mediated Drug Resistance in Bacteria. Curr. Drug Targets 2016, 17, 702–719. [Google Scholar] [CrossRef]
- Pages, J.M.; Masi, M.; Barbe, J. Inhibitors of efflux pumps in Gram-negative bacteria. Trends Mol. Med. 2005, 11, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T.; et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Renau, T.E.; Leger, R.; Filonova, L.; Flamme, E.M.; Wang, M.; Yen, R.; Madsen, D.; Griffith, D.; Chamberland, S.; Dudley, M.N.; et al. Conformationally-restricted analogues of efflux pump inhibitors that potentiate the activity of levofloxacin in Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett. 2003, 13, 2755–2758. [Google Scholar] [CrossRef]
- Yoshida, K.; Nakayama, K.; Ohtsuka, M.; Kuru, N.; Yokomizo, Y.; Sakamoto, A.; Takemura, M.; Hoshino, K.; Kanda, H.; Nitanai, H.; et al. MexAB-OprM specific efflux pump inhibitors in Pseudomonas aeruginosa. Part 7: Highly soluble and in vivo active quaternary ammonium analogue D13-9001, a potential preclinical candidate. Bioorg. Med. Chem. 2007, 15, 7087–7097. [Google Scholar] [CrossRef]
- Opperman, T.J.; Kwasny, S.M.; Kim, H.S.; Nguyen, S.T.; Houseweart, C.; D’Souza, S.; Walker, G.C.; Peet, N.P.; Nikaido, H.; Bowlin, T.L. Characterization of a novel pyranopyridine inhibitor of the AcrAB efflux pump of Escherichia coli. Antimicrob. Agents Chemother. 2014, 58, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Lomovskaya, O.; Bostian, K.A. Practical applications and feasibility of efflux pump inhibitors in the clinic—A vision for applied use. Biochem. Pharmacol. 2006, 71, 910–918. [Google Scholar] [CrossRef]
- Farrell, L.J.; Lo, R.; Wanford, J.J.; Jenkins, A.; Maxwell, A.; Piddock, L.J.V. Revitalizing the drug pipeline: AntibioticDB, an open access database to aid antibacterial research and development. J. Antimicrob. Chemother. 2018, 73, 2284–2297. [Google Scholar] [CrossRef]
- Li, X.Z.; Plesiat, P.; Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, H.Y.; Jamshidi, S.; Sutton, J.M.; Rahman, K.M. Current Advances in Developing Inhibitors of Bacterial Multidrug Efflux Pumps. Curr. Med. Chem. 2016, 23, 1062–1081. [Google Scholar] [CrossRef]
- Schweizer, H.P. Understanding efflux in Gram-negative bacteria: Opportunities for drug discovery. Expert Opin. Drug Discov. 2012, 7, 633–642. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef]
- TAXIS Pharmaceuticals, Inc. Tackling resistance in multidrug-resistant bacterial infections. Biopharma Deal. 2020, 14. Available online: www.nature.com/articles/d43747-020-00935-2 (accessed on 1 November 2021).
- Blankson, G.; Parhi, A.K.; Kaul, M.; Pilch, D.S.; LaVoie, E.J. Structure-activity relationships of potentiators of the antibiotic activity of clarithromycin against Escherichia coli. Eur. J. Med. Chem. 2019, 178, 30–38. [Google Scholar] [CrossRef]
- Blankson, G.A.; Parhi, A.K.; Kaul, M.; Pilch, D.S.; LaVoie, E.J. Advances in the structural studies of antibiotic potentiators against Escherichia coli. Bioorg. Med. Chem. 2019, 27, 3254–3278. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, S.; Li, X.; He, X.; Jian, L. Development of in vitro resistance to fluoroquinolones in Pseudomonas aeruginosa. Antimicrob. Resist. Infect. Control 2020, 9, 124. [Google Scholar] [CrossRef]
- Poole, K. Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 2005, 56, 20–51. [Google Scholar] [CrossRef] [Green Version]
- Kohler, T.; Michea-Hamzehpour, M.; Henze, U.; Gotoh, N.; Curty, L.K.; Pechere, J.C. Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Microbiol. 1997, 23, 345–354. [Google Scholar] [CrossRef]
- Masuda, N.; Sakagawa, E.; Ohya, S.; Gotoh, N.; Tsujimoto, H.; Nishino, T. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3322–3327. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, X.Z.; Poole, K. Fluoroquinolone susceptibilities of efflux-mediated multidrug-resistant Pseudomonas aeruginosa, Stenotrophomonas maltophilia and Burkholderia cepacia. J. Antimicrob. Chemother. 2001, 48, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Avrain, L.; Mertens, P.; Van Bambeke, F. RND efflux pumps in P. aeruginosa: An underestimated resistance mechanism. Antibiot. Susceptibility 2013, 26321, 26–28. [Google Scholar]
- Dean, C.R.; Visalli, M.A.; Projan, S.J.; Sum, P.E.; Bradford, P.A. Efflux-mediated resistance to tigecycline (GAR-936) in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 2003, 47, 972–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fluit, A.C.; Florijn, A.; Verhoef, J.; Milatovic, D. Presence of tetracycline resistance determinants and susceptibility to tigecycline and minocycline. Antimicrob. Agents Chemother. 2005, 49, 1636–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, P.J.; Jacobus, N.V.; Weiss, W.J.; Sum, P.E.; Testa, R.T. In vitro and in vivo antibacterial activities of a novel glycylcycline, the 9-t-butylglycylamido derivative of minocycline (GAR-936). Antimicrob. Agents Chemother. 1999, 43, 738–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, T.; Kok, M.; Michea-Hamzehpour, M.; Plesiat, P.; Gotoh, N.; Nishino, T.; Curty, L.K.; Pechere, J.C. Multidrug efflux in intrinsic resistance to trimethoprim and sulfamethoxazole in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1996, 40, 2288–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Z.; Livermore, D.M.; Nikaido, H. Role of efflux pump(s) in intrinsic resistance of Pseudomonas aeruginosa: Resistance to tetracycline, chloramphenicol, and norfloxacin. Antimicrob. Agents Chemother. 1994, 38, 1732–1741. [Google Scholar] [CrossRef] [Green Version]
- Kohler, T.; Michea-Hamzehpour, M.; Epp, S.F.; Pechere, J.C. Carbapenem activities against Pseudomonas aeruginosa: Respective contributions of OprD and efflux systems. Antimicrob. Agents Chemother. 1999, 43, 424–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, R.E.; Wong, P.G. Compounds which increase the permeability of the Pseudomonas aeruginosa outer membrane. Antimicrob. Agents Chemother. 1984, 26, 48–52. [Google Scholar] [CrossRef] [Green Version]
- Bruchmann, S.; Dotsch, A.; Nouri, B.; Chaberny, I.F.; Haussler, S. Quantitative contributions of target alteration and decreased drug accumulation to Pseudomonas aeruginosa fluoroquinolone resistance. Antimicrob. Agents Chemother. 2013, 57, 1361–1368. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.V.; Nguyen, T.V.; Nguyen, H.T.T.; Le, D.V. Mutations in the gyrA, parC, and mexR genes provide functional insights into the fluoroquinolone-resistant Pseudomonas aeruginosa isolated in Vietnam. Infect. Drug Resist. 2018, 11, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugelberg, E.; Lofmark, S.; Wretlind, B.; Andersson, D.I. Reduction of the fitness burden of quinolone resistance in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2005, 55, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Braz, V.S.; Furlan, J.P.; Fernandes, A.F.; Stehling, E.G. Mutations in NalC induce MexAB-OprM overexpression resulting in high level of aztreonam resistance in environmental isolates of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Garcia-Contreras, R.; Pu, M.; Sheng, L.; Garcia, L.R.; Tomas, M.; Wood, T.K. Quorum quenching quandary: Resistance to antivirulence compounds. ISME J. 2012, 6, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.P.; Xu, Y.H.; Wang, Z.X.; Fang, Y.P.; Shen, J.L. Overexpression of MexAB-OprM efflux pump in carbapenem-resistant Pseudomonas aeruginosa. Arch. Microbiol. 2016, 198, 565–571. [Google Scholar] [CrossRef]
- Choudhury, D.; Ghose, A.; Dhar Chanda, D.; Das Talukdar, A.; Dutta Choudhury, M.; Paul, D.; Maurya, A.P.; Chakravarty, A.; Bhattacharjee, A. Premature Termination of MexR Leads to Overexpression of MexAB-OprM Efflux Pump in Pseudomonas aeruginosa in a Tertiary Referral Hospital in India. PLoS ONE 2016, 11, e0149156. [Google Scholar] [CrossRef]
- Pai, H.; Kim, J.; Kim, J.; Lee, J.H.; Choe, K.W.; Gotoh, N. Carbapenem resistance mechanisms in Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 2001, 45, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, A.; Canton, R.; Campo, P.; Baquero, F.; Blazquez, J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 2000, 288, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Srikumar, R.; Paul, C.J.; Poole, K. Influence of mutations in the mexR repressor gene on expression of the MexA-MexB-oprM multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 2000, 182, 1410–1414. [Google Scholar] [CrossRef] [Green Version]
- Morita, Y.; Sobel, M.L.; Poole, K. Antibiotic inducibility of the MexXY multidrug efflux system of Pseudomonas aeruginosa: Involvement of the antibiotic-inducible PA5471 gene product. J. Bacteriol. 2006, 188, 1847–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purssell, A.; Poole, K. Functional characterization of the NfxB repressor of the mexCD-oprJ multidrug efflux operon of Pseudomonas aeruginosa. Microbiology 2013, 159, 2058–2073. [Google Scholar] [CrossRef] [Green Version]
- Fetar, H.; Gilmour, C.; Klinoski, R.; Daigle, D.M.; Dean, C.R.; Poole, K. mexEF-oprN multidrug efflux operon of Pseudomonas aeruginosa: Regulation by the MexT activator in response to nitrosative stress and chloramphenicol. Antimicrob. Agents Chemother. 2011, 55, 508–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paixao, L.; Rodrigues, L.; Couto, I.; Martins, M.; Fernandes, P.; de Carvalho, C.C.; Monteiro, G.A.; Sansonetty, F.; Amaral, L.; Viveiros, M. Fluorometric determination of ethidium bromide efflux kinetics in Escherichia coli. J. Biol. Eng. 2009, 3, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. Pharmacokinetics and in vivo antistaphylococcal efficacy of TXY541, a 1-methylpiperidine-4-carboxamide prodrug of PC190723. Biochem. Pharmacol. 2013, 86, 1699–1707. [Google Scholar] [CrossRef]
- Keepers, T.R.; Gomez, M.; Celeri, C.; Nichols, W.W.; Krause, K.M. Bactericidal activity, absence of serum effect, and time-kill kinetics of ceftazidime-avibactam against beta-lactamase-producing Enterobacteriaceae and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 5297–5305. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
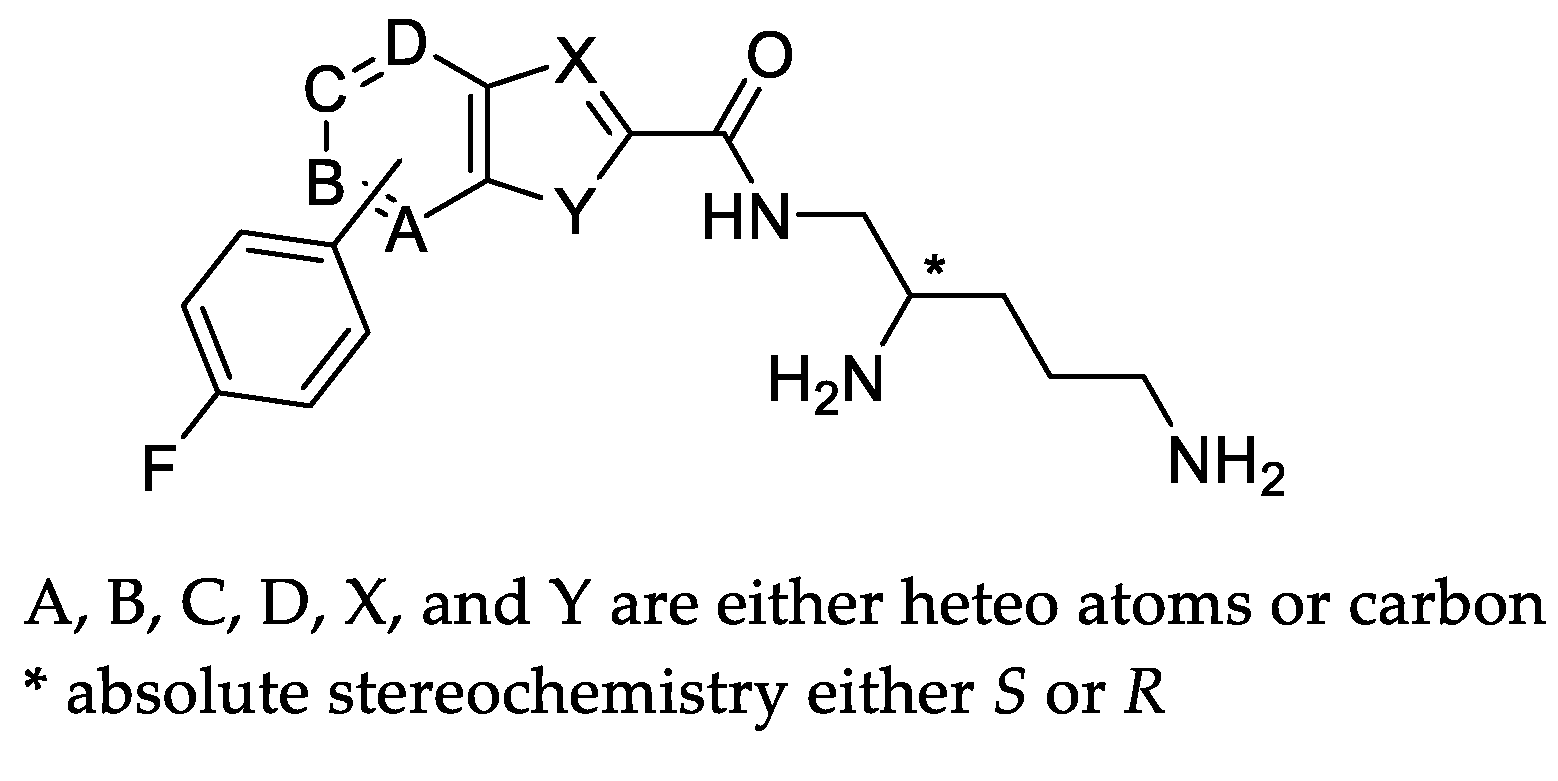
| Compound | X | Y | B | * Stereo | Compound | X | Y | C | * Stereo | |
| 6a | N | S | CH | S | 6b | N | S | CH | S | |
| 6c | N | NH | CH | S | 6f | CH | O | CH | S | |
| 6d | N | NH | CH | R | 6h | CH | S | CH | S | |
| 6e | CH | O | CH | S | 6j (TXA01182) | CH | NH | CH | S | |
| 6g | CH | S | CH | S | 6l | CH | NH | CH | R | |
| 6i | CH | NH | CH | S | 6n | CH | NH | N | S | |
| 6k | CH | NH | CH | R | ||||||
| 6m | CH | NH | N | S | ||||||
| Compound | 6a | 6b | 6c | 6d | 6e | 6f | 6g | 6h | 6i | 6j | 6k | 6l | 6m | 6n |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC of EPI (µg/mL) | 100 | >100 | 100 | >100 | 100 | >100 | 25 | 25 | 100 | 100 | >100 | 100 | >100 | >100 |
| LVX MIC in the presence of EPI (µg/mL) | 1 | 1 | 0.50 | 0.50 | 1 | 0.25 | 0.03 | 0.25 | 0.25 | 0.13 | 0.50 | 0.13 | 1 | 1 |
| Fold Difference | 1 | 1 | 2 | 2 | 1 | 4 | 32 | 4 | 4 | 8 | 2 | 8 | 1 | 1 |
| Antimicrobial | MICs (µg/mL) | Fold Difference | |
|---|---|---|---|
| Alone | + TXA01182 (6.25 µg/mL) | ||
| Aztreonam | 8 | 2 | 4 |
| Cefepime | 2 | 1 | 2 |
| Ceftazidime | 2 | 1 | 2 |
| Azithromycin | 64 | 32 | 2 |
| Ciprofloxacin | 0.25 | 0.125 | 2 |
| Moxifloxacin | 2 | 0.063 | 32 |
| Levofloxacin | 1 | 0.125 | 8 |
| Cotrimoxazole | >256 | 16 | >16 |
| Doxycycline | 32 | 2 | 16 |
| Minocycline | 32 | 1 | 32 |
| Tigecycline | 16 | 4 | 4 |
| Chloramphenicol | >256 | 32 | >8 |
| Imipenem # | 4 | 4 | 1 |
| Gentamicin # | 2 | 2 | 1 |
| Strain | Levofloxacin MIC (μg/mL), (Fold Difference) | Resistance Mechanisms | ||||
|---|---|---|---|---|---|---|
| No EPI | + TXA01182 (6.25 μg/mL) | + MC-04,124 (6.25 μg/mL) | + PAβN (50 μg/mL) | + CCCP (12.5 μg/mL) | ||
| AR-0229 | 64 | 4, (16) | 64, (1) | 64, (1) | 64, (1) | gyrA-T83I, nalC-G71E, mexR-V126Q, OXA-50, PAO |
| AR-0239 | 64 | 8, (8) | 64, (1) | 8, (8) | 64, (1) | gyrA-T83I, nalC-G71E, mexR-V126Q, aac(6’)-IIa, aadB, aph(3’)-Ic, cmlA1, dfrB5, GES-1, OXA-10, OXA-50, strA, strB, tet(G), VIM-11 |
| AR-0244 | 64 | 8, (8) | 64, (1) | 64, (1) | 64, (1) | gyrA-T133H, nalC-G71E, mexR-V126Q, OXA-50 |
| AR-0246 | 64 | 8, (8) | 64, (1) | 64, (1) | 64, (1) | gyrA-T83I, nalC-G71E, mexR-V126Q, aadB, NDM-1, OXA-10, OXA-50, PAO, rmtD2, tet(G), VEB-1 |
| AR-0249 | 64 | 4, (16) | 64, (1) | 8, (8) | 64, (1) | gyrA-T83I, nalC-G71E, aac(3)-Id, aadA2, cmlA1, dfrB5, OXA-4, OXA-50, PAO, tet(G), VIM-2 |
| AR-0264 | 64 | 4, (16) | 64, (1) | 64, (1) | 64, (1) | gyrA-D87Y, nalC-G71E, OXA-50, PAO |
| AR-0232 | 8 | 0.5, (16) | ND | ND | 8, (1) | gyrA-T83I, nalC-G71E, mexR-V126Q, aadA6, OXA-50, PAO, strA, strB, sul1, tet(C) |
| AR-0234 | 8 | 0.25, (32) | ND | ND | 8, (1) | gyrA-T83I, nalC-G71E, mexR-V126Q, aadA6, OXA-50, PAO, strA, strB, tet(C) |
| Strain | TXA01182 (6.25 μg/mL) | Levofloxacin (4 μg/mL) | Levofloxacin (4 μg/mL) + TXA01182 (6.25 μg/mL) |
|---|---|---|---|
| P. aeruginosa ATCC 27853 | 0.73 | 7.44 × 10−8 | <1.30 × 10−10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Y.; Rosado-Lugo, J.D.; Zhang, Y.; Datta, P.; Sun, Y.; Cao, Y.; Banerjee, A.; Parhi, A.K. Evaluation of Heterocyclic Carboxamides as Potential Efflux Pump Inhibitors in Pseudomonas aeruginosa. Antibiotics 2022, 11, 30. https://doi.org/10.3390/antibiotics11010030
Yuan Y, Rosado-Lugo JD, Zhang Y, Datta P, Sun Y, Cao Y, Banerjee A, Parhi AK. Evaluation of Heterocyclic Carboxamides as Potential Efflux Pump Inhibitors in Pseudomonas aeruginosa. Antibiotics. 2022; 11(1):30. https://doi.org/10.3390/antibiotics11010030
Chicago/Turabian StyleYuan, Yi, Jesus D. Rosado-Lugo, Yongzheng Zhang, Pratik Datta, Yangsheng Sun, Yanlu Cao, Anamika Banerjee, and Ajit K. Parhi. 2022. "Evaluation of Heterocyclic Carboxamides as Potential Efflux Pump Inhibitors in Pseudomonas aeruginosa" Antibiotics 11, no. 1: 30. https://doi.org/10.3390/antibiotics11010030