
Whole-Genome Sequencing Reveals Recent Transmission of Multidrug-Resistant Mycobacterium tuberculosis CAS1-Kili Strains in Lusaka, Zambia

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
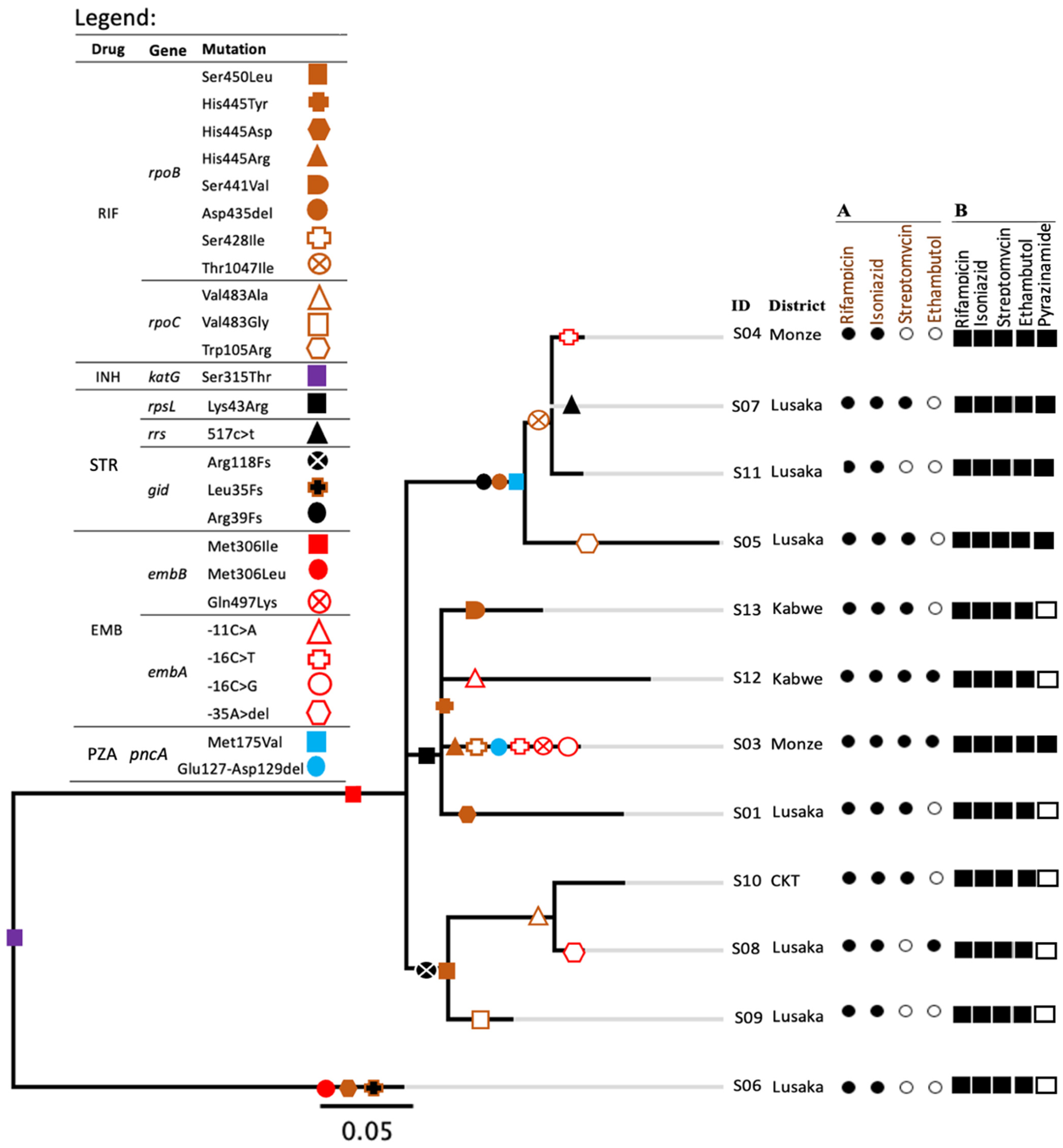
2.1. Cluster Analysis
2.2. Resistance Patterns and Phylogeny
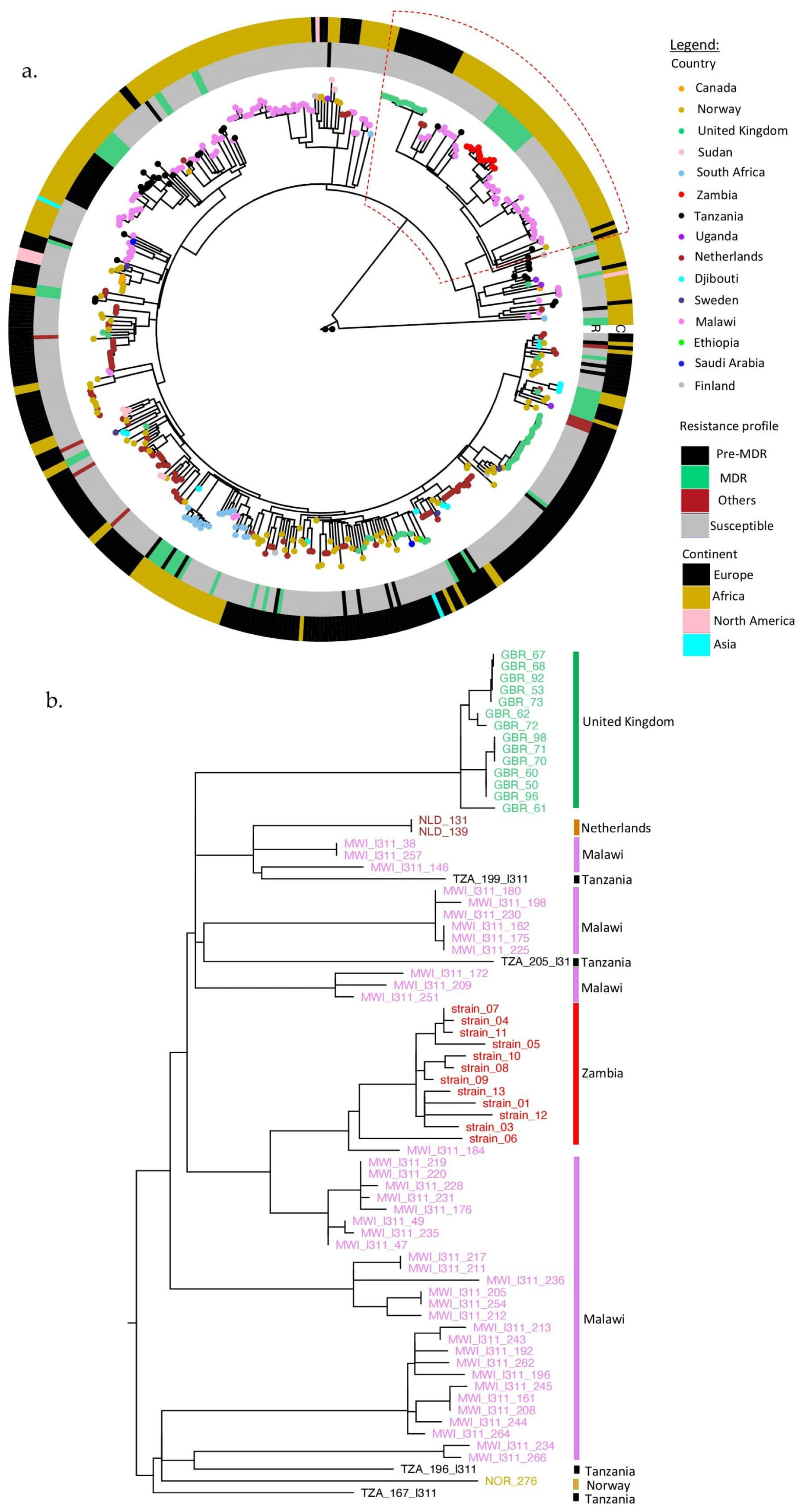
2.3. Phylogenetic Assessment of Global Sub-Lineage SIT21/CAS1-Kili (L3.1.1)
3. Discussion
4. Materials and Methods
4.1. Study Samples
4.2. Culturing and Drug Susceptibility Testing
4.3. DNA Extraction and Genotyping
4.4. Whole-Genome Sequencing Analysis
4.5. Phylogenetic Assessment of Global Sub-Lineage SIT21/CAS1-Kili (L3.1.1)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2020; WHO: Geneva, Switzerland, 2020; ISBN 9789240013131. [Google Scholar]
- UNAIDS. Tuberculosis and Hiv Tb Is the Leading Cause of Death; UNAIDS: Geneva, Switzerland, 2018; Volume 54, pp. 2020–2235. [Google Scholar]
- Chihota, V.N.; Niehaus, A.; Streicher, E.M.; Wang, X.; Sampson, S.L.; Mason, P.; Källenius, G.; Mfinanga, S.G.; Pillay, M.; Klopper, M.; et al. Geospatial distribution of Mycobacterium tuberculosis genotypes in Africa. PLoS ONE 2018, 13, e0200632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, M.B.; Shockey, A.; Zarley, A.; Aylward, W.; Eldholm, V.; Kitchen, A.; Pepperell, C.S. Lineage specific histories of Mycobacterium tuberculosis dispersal in Africa and Eurasia. Mol. Ecol. 2019, 28, 3241–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagneux, S.; DeRiemer, K.; Van, T.; Kato-Maeda, M.; de Jong, B.C.; Narayanan, S.; Nicol, M.; Niemann, S.; Kremer, K.; Gutierrez, M.C.; et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2006, 103, 2869–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supply, P.; Allix, C.; Lesjean, S.; Cardoso-Oelemann, M.; Rüsch-Gerdes, S.; Willery, E.; Savine, E.; de Haas, P.; van Deutekom, H.; Roring, S.; et al. Proposal for Standardization of Optimized Mycobacterial Interspersed Repetitive Unit – Variable-Number Tandem Repeat Typing of Mycobacterium tuberculosis. J. Clin. Microbiol. 2006, 44, 4498–4510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamerbeek, J.; Schouls, L.; Kolk, A.; van Agterveld, M.; van Soolingen, D.; Kuijper, S.; Bunschoten, A.; Molhuizen, H.; Shaw, R.; Goyal, M.; et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 1997, 35, 907–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Embden, J.D.A.; Cave, M.D.; Crawford, J.T.; Dale, J.W.; Eisenach, K.D.; Gicquel, B.; Hermans, P.; Martin, C.; McAdam, R.; Shinnick, T.M.; et al. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: Recommendations for a standardized methodology. J. Clin. Microbiol. 1993, 31, 406–409. [Google Scholar] [CrossRef] [Green Version]
- Easterbrook, P.J.; Gibson, A.; Murad, S.; Lamprecht, D.; Ives, N.; Ferguson, A.; Lowe, O.; Mason, P.; Ndudzo, A.; Taziwa, A.; et al. High rates of clustering of strains causing tuberculosis in Harare, Zimbabwe: A molecular epidemiological study. J. Clin. Microbiol. 2004, 42, 4536–4544. [Google Scholar] [CrossRef] [Green Version]
- Hamblion, E.L.; Le Menach, A.; Anderson, L.F.; Lalor, M.K.; Brown, T.; Abubakar, I.; Anderson, C.; Maguire, H.; Anderson, S.R.; Watson, J.; et al. Recent TB transmission, clustering and predictors of large clusters in London, 2010–2012: Results from first 3-years of universal MIRU-VNTR strain typing. Thorax 2016, 71, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Mao, X.; Wang, J.; Pan, H. Clustering and recent transmission of Mycobacterium tuberculosis in a Chinese population. Infect. Drug Resist. 2018, 11, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Roetzer, A.; Diel, R.; Kohl, T.A.; Rückert, C.; Nübel, U.; Blom, J.; Wirth, T.; Jaenicke, S.; Schuback, S.; Rüsch-Gerdes, S.; et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: A longitudinal molecular epidemiological study. PLoS Med. 2013, 10, e1001387. [Google Scholar] [CrossRef]
- Luo, T.; Yang, C.; Peng, Y.; Lu, L.; Sun, G.; Wu, J.; Jin, X.; Hong, J.; Li, F.; Mei, J.; et al. Whole-genome sequencing to detect recent transmission of Mycobacterium tuberculosis in settings with a high burden of tuberculosis. Tuberculosis (Edinb). 2014, 94, 434–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, I.; Cerezo, F. Bioinformatic Analysis of Mycobacterium tuberculosis whole genome data. Ph.D. Thesis, London School of Hygiene & Tropical Medicine, London, UK, 2015. [Google Scholar]
- Walker, T.M.; Ip, C.L.C.; Harrell, R.H.; Evans, J.T.; Kapatai, G.; Dedicoat, M.J.; Eyre, D.W.; Wilson, D.J.; Hawkey, P.M.; Crook, D.W.; et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet. Infect. Dis. 2013, 13, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Feliciano, C.S.; Namburete, E.I.; Rodrigues Plaça, J.; Peronni, K.; Dippenaar, A.; Warren, R.M.; Silva, W.A.; Bollela, V.R. Accuracy of whole genome sequencing versus phenotypic (MGIT) and commercial molecular tests for detection of drug-resistant Mycobacterium tuberculosis isolated from patients in Brazil and Mozambique. Tuberculosis 2018, 110, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Lalor, M.; Casali, N.; Walker, T.; Anderson, L.; Davidson, J.; Ratna, N.; Mullarkey, C.; Gent, M.; Foster, K.; Brown, T.; et al. The use of whole-genome sequencing in cluster investigation of an MDR-TB outbreak. Eur. Respir. J. 2018, 51, 1702313. [Google Scholar] [CrossRef]
- Chizimu, J.Y.; Solo, E.S.; Bwalya, P.; Kapalamula, T.F.; Akapelwa, M.L.; Lungu, P.; Shrestha, D.; Fukushima, Y.; Mukinka, V.; Thapa, J.; et al. Genetic Diversity and Transmission of Multidrug Resistant Mycobacterium tuberculosis strains in Lusaka, Zambia. Int. J. Infect. Dis. 2021, 114, 142–150. [Google Scholar] [CrossRef]
- Solo, E.S.; Suzuki, Y.; Kaile, T.; Bwalya, P.; Lungu, P.; Chizimu, J.Y.; Shah, Y.; Nakajima, C. Characterization of Mycobacterium tuberculosis genotypes and their correlation to multidrug resistance in Lusaka, Zambia. Int. J. Infect. Dis. 2021, 102, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I. The biological cost of mutational antibiotic resistance: Any practical conclusions? Curr. Opin. Microbiol. 2006, 9, 461–465. [Google Scholar] [CrossRef]
- Asare, P.; Otchere, I.D.; Bedeley, E.; Brites, D.; Loiseau, C.; Baddoo, N.A.; Asante-Poku, A.; Osei-Wusu, S.; Prah, D.A.; Borrell, S.; et al. Whole Genome Sequencing and Spatial Analysis Identifies Recent Tuberculosis Transmission Hotspots in Ghana. Front. Med. 2020, 7, 161. [Google Scholar] [CrossRef]
- Metcalfe, J.Z.; Kim, E.Y.; Lin, S.-Y.G.; Cattamanchi, A.; Oh, P.; Flood, J.; Hopewell, P.C.; Kato-Maeda, M. Determinants of Multidrug-Resistant Tuberculosis Clusters, California, USA, 2004–2007. Emerg. Infect. Dis. J. 2010, 16, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.C.C.; Leung, C.C.; Kam, K.M.; Yew, W.W.; Chang, K.C.; Leung, W.M.; Tam, C.M. Transmission of multidrug-resistant and extensively drug-resistant tuberculosis in a metropolitan city. Eur. Respir. J. 2013, 41, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Comas, I.; Borrell, S.; Roetzer, A.; Rose, G.; Malla, B.; Kato-Maeda, M.; Galagan, J.; Niemann, S.; Gagneux, S. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 2011, 44, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Xiao, T.Y.; Liu, H.C.; Zhao, X.Q.; Liu, Z.G.; Li, Y.N.; Zeng, H.; Zhao, L.L.; Wan, K.L. Mutations within embCAB Are Associated with Variable Level of Ethambutol Resistance in Mycobacterium tuberculosis Isolates from China. Antimicrob. Agents Chemother. 2017, 62, e01279-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Chen, R.; Lin, S.; Lu, Y.; Liu, H.; Li, G.; Liu, Z.; Zhao, X.; Zhao, L.; Wan, K.-L. Detecting Ethambutol Resistance in Mycobacterium tuberculosis Isolates in China: A Comparison Between Phenotypic Drug Susceptibility Testing Methods and DNA Sequencing of embAB. Front. Microbiol. 2020, 11, 781. [Google Scholar] [CrossRef]
- Bakuła, Z.; Napiórkowska, A.; Bielecki, J.; Augustynowicz-Kopeć, E.; Zwolska, Z.; Jagielski, T. Mutations in the embB Gene and Their Association with Ethambutol Resistance in Multidrug-Resistant Mycobacterium tuberculosis Clinical Isolates from Poland. BioMed Res. Int. 2013, 2013, 167954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safi, H.; Sayers, B.; Hazbón, M.H.; Alland, D. Transfer of embB Codon 306 Mutations into Clinical Mycobacterium tuberculosis Strains Alters Susceptibility to Ethambutol, Isoniazid, and Rifampin. Antimicrob. Agents Chemother. 2008, 52, 2027–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, L.M.; Salfinger, M.; Clobridge, A.; Dormandy, J.; Mirabello, L.; Polletta, V.L.; Sanic, A.; Sinyavskiy, O.; Larsen, S.C.; Driscoll, J.; et al. Phenotypic and Molecular Characterization of Mycobacterium tuberculosis Isolates Resistant to both Isoniazid and Ethambutol. Antimicrob. Agents Chemother. 2005, 49, 2218–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazbón, M.H.; Valle, M.B.; del Guerrero, M.I.; Varma-Basil, M.; Filliol, I.; Cavatore, M.; Colangeli, R.; Safi, H.; Billman-Jacobe, H.; Lavender, C.; et al. Role of embB Codon 306 Mutations in Mycobacterium tuberculosis Revisited: A Novel Association with Broad Drug Resistance and IS6110 Clustering Rather than Ethambutol Resistance. Antimicrob. Agents Chemother. 2005, 49, 3794–3802. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Shen, G.; Wu, J.; Gui, X.; Li, X.; Mei, J.; DeRiemer, K.; Gao, Q. Association between embB Codon 306 Mutations and Drug Resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 2618–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuaib, Y.A.; Khalil, E.A.G.; Wieler, L.H.; Schaible, U.E.; Bakheit, M.A.; Mohamed-Noor, S.E.; Abdalla, M.A.; Kerubo, G.; Andres, S.; Hillemann, D.; et al. Mycobacterium tuberculosis Complex Lineage 3 as Causative Agent of Pulmonary Tuberculosis, Eastern Sudan. Emerg. Infect. Dis. 2020, 26, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.Y.; Lee, J.S.; Kwak, H.K.; Via, L.E.; Boshoff, H.I.M.; Barry, C.E. Mutations in gidB Confer Low-Level Streptomycin Resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 2515–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, A.; Sander, P.; Schaper, K.J.; Scholz, M.; Böttger, E.C. Correlation of molecular resistance mechanisms and phenotypic resistance levels in streptomycin-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1996, 40, 2452–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bwalya, P.; Yamaguchi, T.; Solo, E.S.; Chizimu, J.Y.; Mbulo, G.; Nakajima, C.; Suzuki, Y. Characterization of Mutations Associated with Streptomycin Resistance in Multidrug-Resistant Mycobacterium tuberculosis in Zambia. Antibiot. 2021, 10, 1169. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, C.; Xiang, L.; Pi, R.; Guo, Z.; Zheng, C.; Li, S.; Zhao, Y.; Tang, K.; Luo, M.; et al. Characterization of mutations in streptomycin-resistant Mycobacterium tuberculosis isolates in Sichuan, China and the association between Beijing-lineage and dual-mutation in gidB. Tuberculosis 2016, 96, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cancino-Muñoz, I.; Torres-Puente, M.; Villamayor, L.M.; Borrá, R.; Borrás-Máñez, M.; Bosque, M.; Camarena, J.J.; Colomer-Roig, E.; Colomina, J.; et al. High-resolution mapping of tuberculosis transmission: Whole genome sequencing and phylogenetic modelling of a cohort from Valencia Region, Spain. PLoS Med. 2019, 16, e1002961. [Google Scholar] [CrossRef] [Green Version]
- Alaridah, N.; Hallbäck, E.T.; Tångrot, J.; Winqvist, N.; Sturegård, E.; Florén-Johansson, K.; Jönsson, B.; Tenland, E.; Welinder-Olsson, C.; Medstrand, P.; et al. Transmission dynamics study of tuberculosis isolates with whole genome sequencing in southern Sweden. Sci. Rep. 2019, 9, 4931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casper, C.; Singh, S.P.; Rave, S.; Daley, C.L.; Schecter, G.S.; Riley, L.W.; Kreiswirth, B.N.; Small, P.M. The transcontinental transmission of tuberculosis: A molecular epidemiological assessment. Am. J. Public Health 1996, 86, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Alyamani, E.J.; Marcus, S.A.; Ramirez-Busby, S.M.; Hansen, C.; Rashid, J.; El-kholy, A.; Spalink, D.; Valafar, F.; Almehdar, H.A.; Fatani, A.J.; et al. Genomic analysis of the emergence of drug-resistant strains of Mycobacterium tuberculosis in the Middle East. Sci. Rep. 2019, 9, 4474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, T.M.; Merker, M.; Knoblauch, A.M.; Helbling, P.; Schoch, O.D.; van der Werf, M.J.; Kranzer, K.; Fiebig, L.; Kröger, S.; Haas, W.; et al. A cluster of multidrug-resistant Mycobacterium tuberculosis among patients arriving in Europe from the Horn of Africa: A molecular epidemiological study. Lancet Infect. Dis. 2018, 18, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Heusden, P.; van Gladman, S.; Lose, T. M.tuberculosis Variant Analysis (Galaxy Training Materials). Available online: https://training.galaxyproject.org/training-material/topics/variant-analysis/tutorials/tb-variant-analysis/tutorial.html (accessed on 14 October 2021).
- Batut, B.; Hiltemann, S.; Bagnacani, A.; Baker, D.; Bhardwaj, V.; Blank, C.; Bretaudeau, A.; Brillet-Guéguen, L.; Čech, M.; Chilton, J.; et al. Community-Driven Data Analysis Training for Biology. Cell Syst. 2018, 6, 752–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Seemann. T Snippy: Rapid Haploid Variant Calling and Core Genome Alignment. Available online: https://github.com/tseemann/snippy (accessed on 23 August 2021).
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2014, 43, e15. [Google Scholar] [CrossRef] [Green Version]
- Phelan, J.E.; O’Sullivan, D.M.; Machado, D.; Ramos, J.; Oppong, Y.E.A.; Campino, S.; O’Grady, J.; McNerney, R.; Hibberd, M.L.; Viveiros, M.; et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 2019, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Coll, F.; McNerney, R.; Preston, M.D.; Guerra-Assunção, J.A.; Warry, A.; Hill-Cawthorne, G.; Mallard, K.; Nair, M.; Miranda, A.; Alves, A.; et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 2015, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Feuerriegel, S.; Schleusener, V.; Beckert, P.; Kohl, T.A.; Miotto, P.; Cirillo, D.M.; Cabibbe, A.M.; Niemann, S.; Fellenberg, K.; Carroll, K.C. PhyResSE: A Web Tool Delineating Mycobacterium tuberculosis Antibiotic Resistance and Lineage from Whole-Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 1908–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katale, B.Z.; Mbelele, P.M.; Lema, N.A.; Campino, S.; Mshana, S.E.; Rweyemamu, M.M.; Phelan, J.E.; Keyyu, J.D.; Majigo, M.; Mbugi, E.V.; et al. Whole genome sequencing of Mycobacterium tuberculosis isolates and clinical outcomes of patients treated for multidrug-resistant tuberculosis in Tanzania. BMC Genom. 2020, 21, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Yu, G.; Lam, T.T.-Y.; Zhu, H.; Guan, Y. Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using Ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. GGTREE: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Team, Rs. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA; Available online: https://www.rstudio.com/ (accessed on 27 July 2021).
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Strain ID | SNP Difference 1 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S01 | S03 | S13 | S08 | S10 | S09 | S12 | S04 | S07 | S11 | S05 | S06 | ≤5 | ≤12 | ≥13 | |
| S01 | 9 | 8 | 11 | 12 | 9 | 11 | 11 | 10 | 11 | 14 | 24 | 0 | 9 | 2 | |
| S03 | 9 | 7 | 10 | 11 | 8 | 10 | 10 | 9 | 10 | 13 | 23 | 0 | 9 | 2 | |
| S13 | 8 | 7 | 9 | 10 | 7 | 9 | 9 | 8 | 9 | 12 | 22 | 0 | 10 | 1 | |
| S08 | 11 | 10 | 9 | 3 | 5 | 12 | 10 | 9 | 10 | 13 | 23 | 2 | 9 | 2 | |
| S10 | 12 | 11 | 10 | 3 | 6 | 13 | 11 | 10 | 11 | 14 | 24 | 1 | 8 | 3 | |
| S09 | 9 | 8 | 7 | 5 | 6 | 10 | 8 | 7 | 8 | 11 | 21 | 1 | 10 | 1 | |
| S12 | 11 | 10 | 9 | 12 | 13 | 10 | 12 | 11 | 12 | 15 | 25 | 0 | 8 | 3 | |
| S04 | 11 | 10 | 9 | 10 | 11 | 8 | 12 | 1 | 2 | 7 | 23 | 2 | 8 | 1 | |
| S07 | 10 | 9 | 8 | 9 | 10 | 7 | 11 | 1 | 1 | 6 | 22 | 2 | 10 | 1 | |
| S11 | 11 | 10 | 9 | 10 | 11 | 8 | 12 | 2 | 1 | 7 | 23 | 2 | 10 | 1 | |
| S05 | 14 | 13 | 12 | 13 | 14 | 11 | 15 | 7 | 6 | 7 | 26 | 0 | 5 | 6 | |
| S06 | 24 | 23 | 22 | 23 | 24 | 21 | 25 | 23 | 22 | 23 | 26 | 0 | 0 | 11 | |
 ≤ 5 SNPs;
≤ 5 SNPs;  ≤ 12 SNPs;
≤ 12 SNPs;  ≥ 13 SNPs. 1 Number of strains differing by ≤5, ≤12, and ≥13 SNPs to an individual strain. The letter S before the ID stands for strain. Boxes are highlighted according to the SNP differences as shown in the legend.
≥ 13 SNPs. 1 Number of strains differing by ≤5, ≤12, and ≥13 SNPs to an individual strain. The letter S before the ID stands for strain. Boxes are highlighted according to the SNP differences as shown in the legend.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chizimu, J.Y.; Solo, E.S.; Bwalya, P.; Tanomsridachchai, W.; Chambaro, H.; Shawa, M.; Kapalamula, T.F.; Lungu, P.; Fukushima, Y.; Mukonka, V.; et al. Whole-Genome Sequencing Reveals Recent Transmission of Multidrug-Resistant Mycobacterium tuberculosis CAS1-Kili Strains in Lusaka, Zambia. Antibiotics 2022, 11, 29. https://doi.org/10.3390/antibiotics11010029
Chizimu JY, Solo ES, Bwalya P, Tanomsridachchai W, Chambaro H, Shawa M, Kapalamula TF, Lungu P, Fukushima Y, Mukonka V, et al. Whole-Genome Sequencing Reveals Recent Transmission of Multidrug-Resistant Mycobacterium tuberculosis CAS1-Kili Strains in Lusaka, Zambia. Antibiotics. 2022; 11(1):29. https://doi.org/10.3390/antibiotics11010029
Chicago/Turabian StyleChizimu, Joseph Yamweka, Eddie Samuneti Solo, Precious Bwalya, Wimonrat Tanomsridachchai, Herman Chambaro, Misheck Shawa, Thoko Flav Kapalamula, Patrick Lungu, Yukari Fukushima, Victor Mukonka, and et al. 2022. "Whole-Genome Sequencing Reveals Recent Transmission of Multidrug-Resistant Mycobacterium tuberculosis CAS1-Kili Strains in Lusaka, Zambia" Antibiotics 11, no. 1: 29. https://doi.org/10.3390/antibiotics11010029