
Effect of Cephalosporin Treatment on the Microbiota and Antibiotic Resistance Genes in Feces of Dairy Cows with Clinical Mastitis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
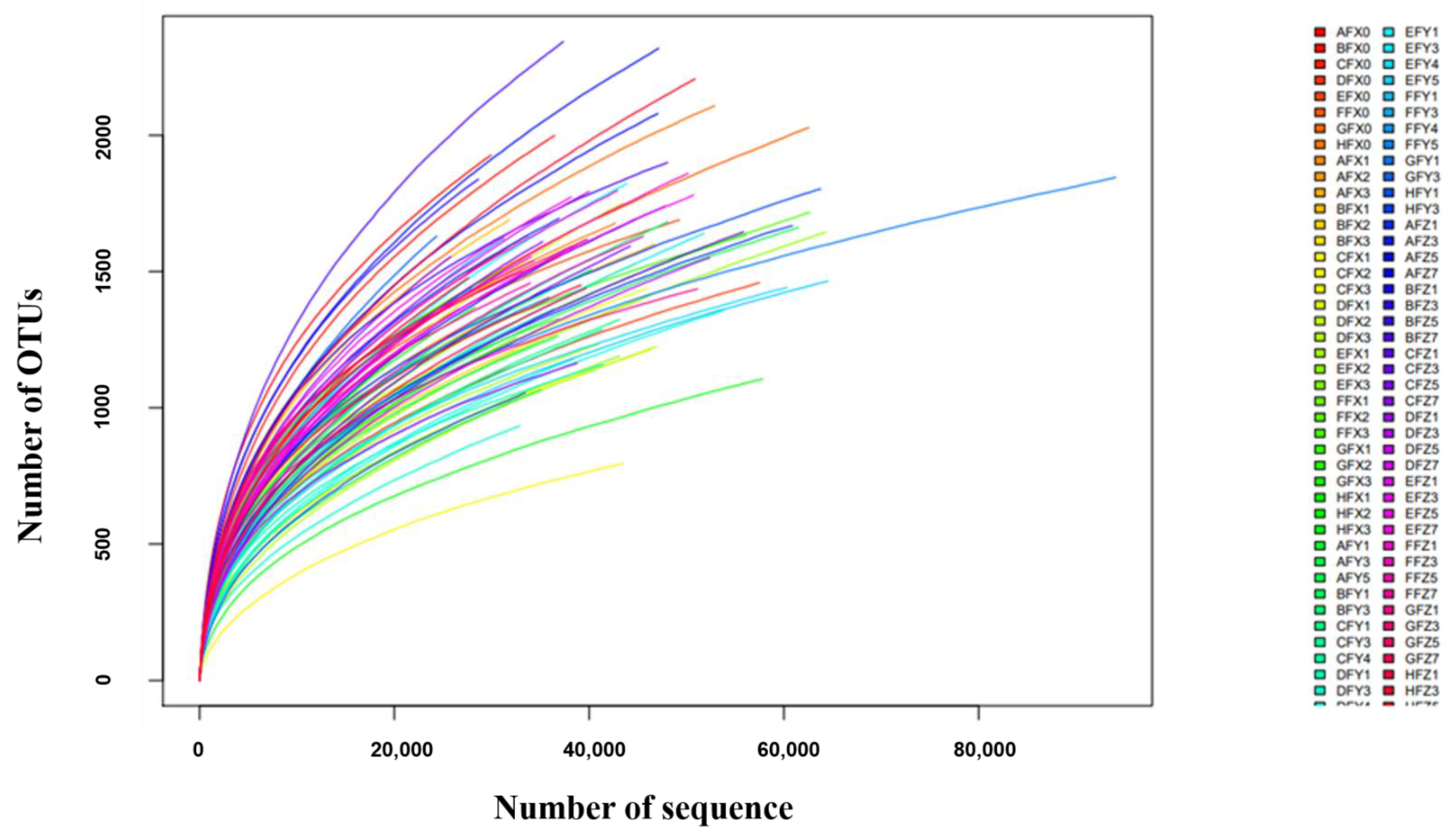
2.1. Animal Weight Gain, and 16S rRNA Gene Sequencing Overview
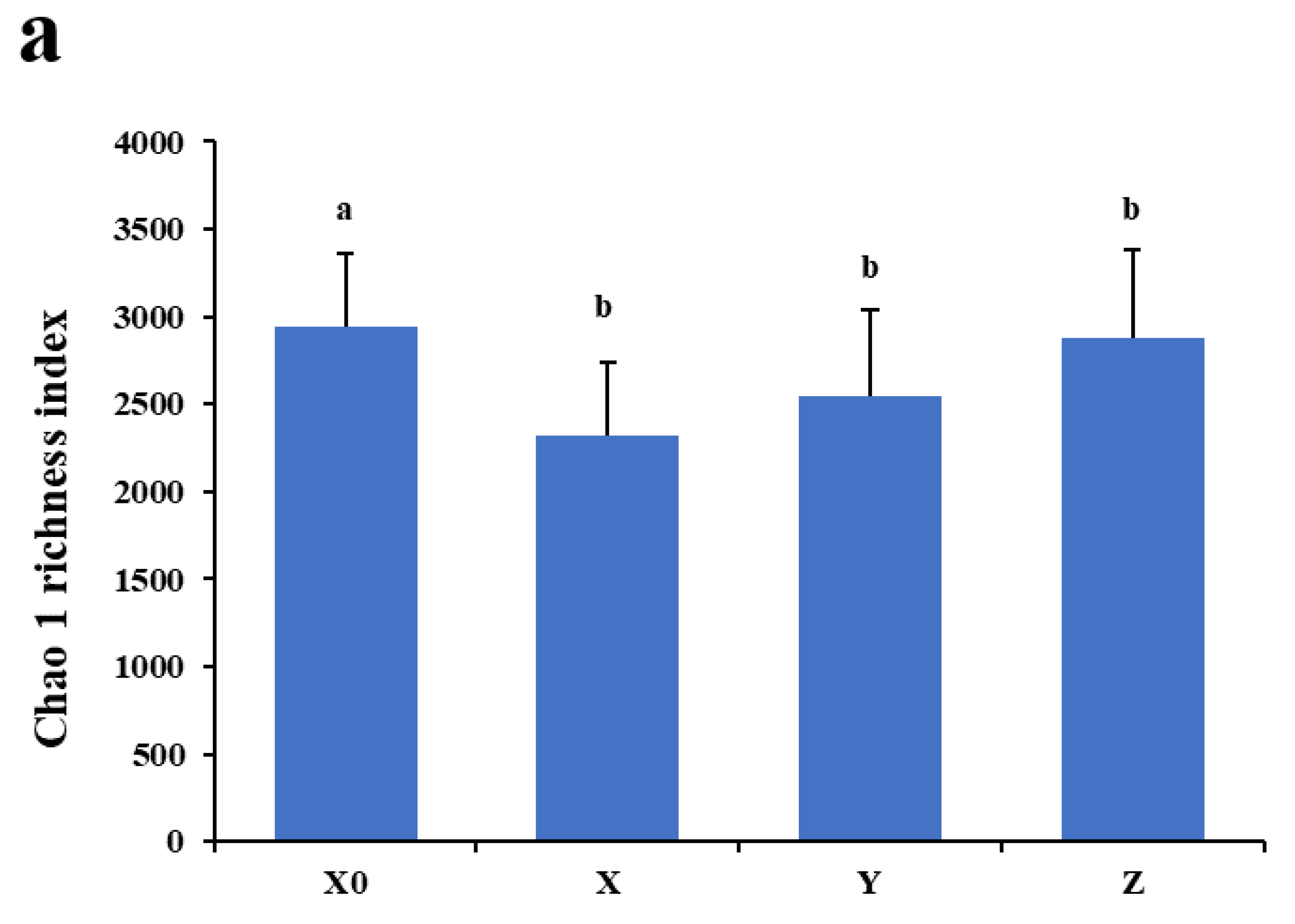
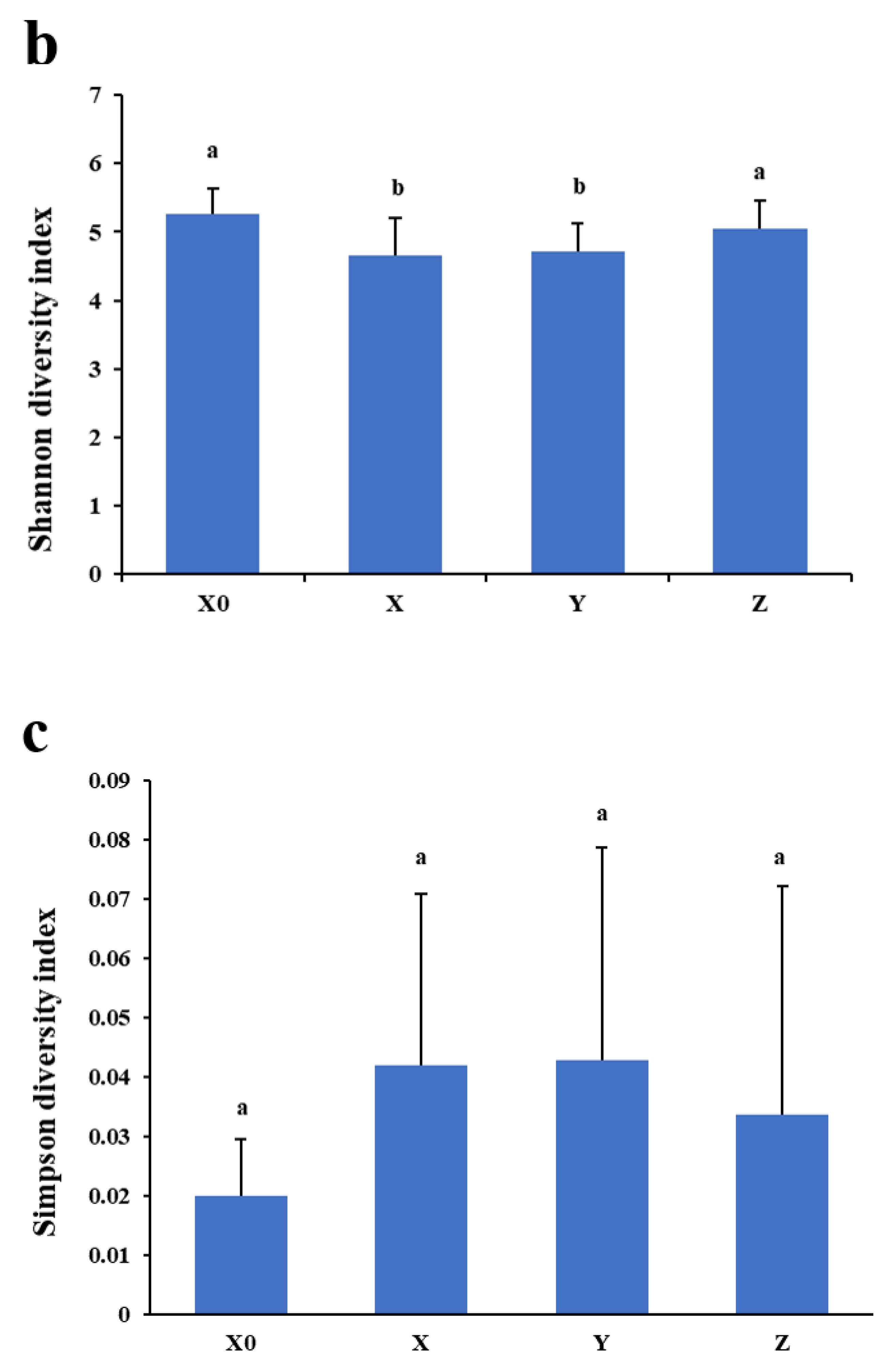
2.2. α Diversity
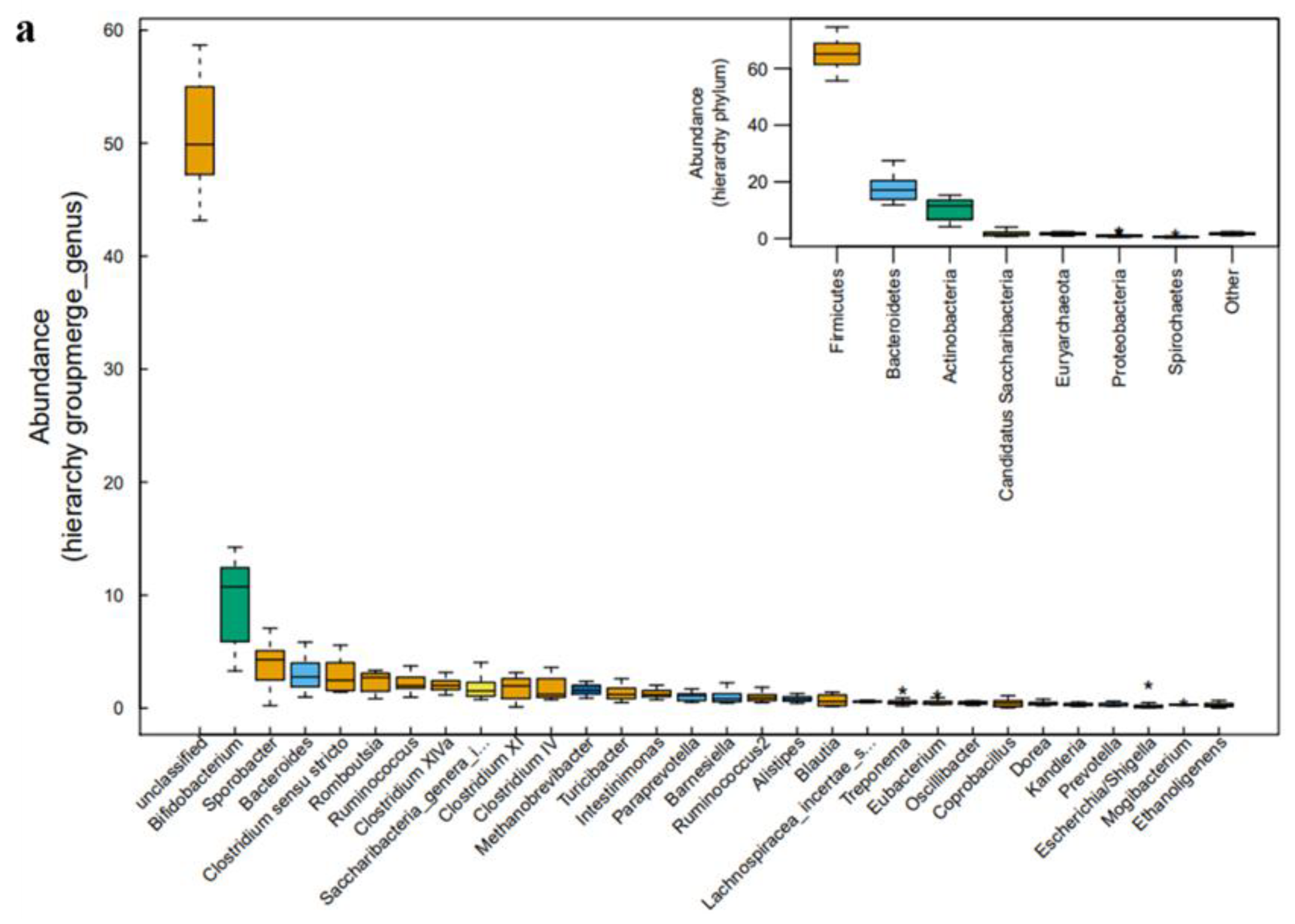
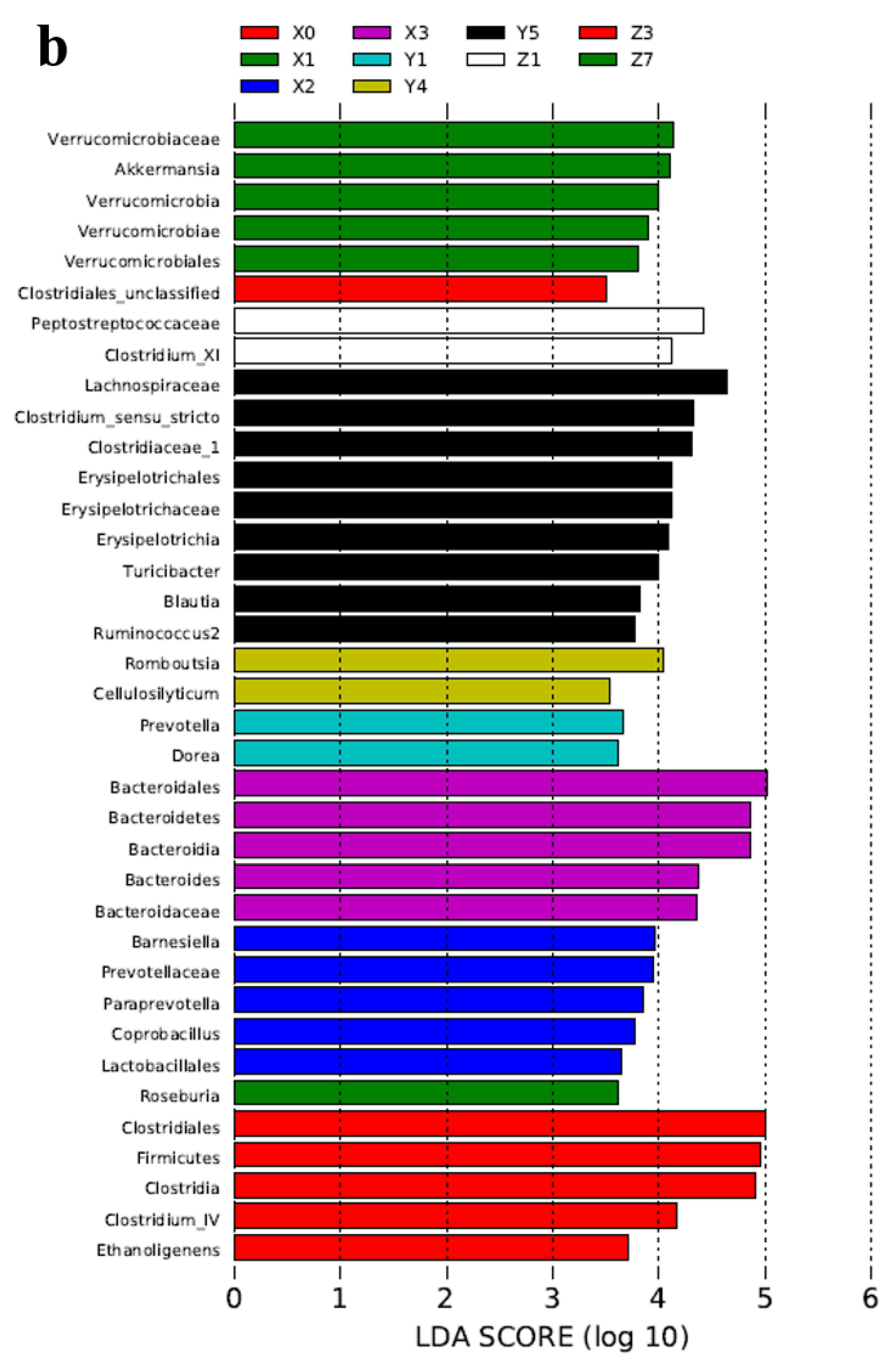
2.3. Microbial Community Analysis
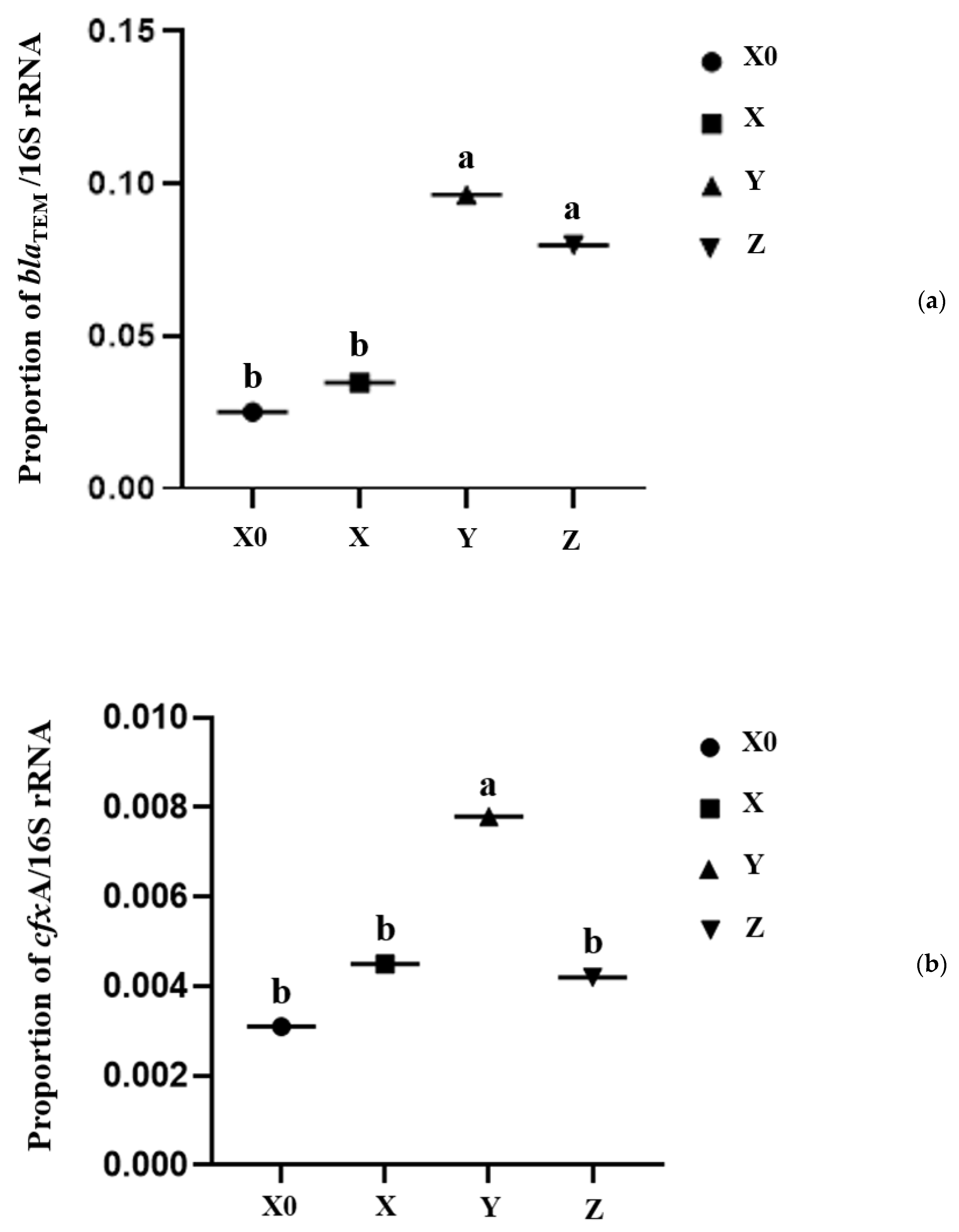
2.4. Antibiotic Resistance Genes
3. Discussion
4. Materials and Methods
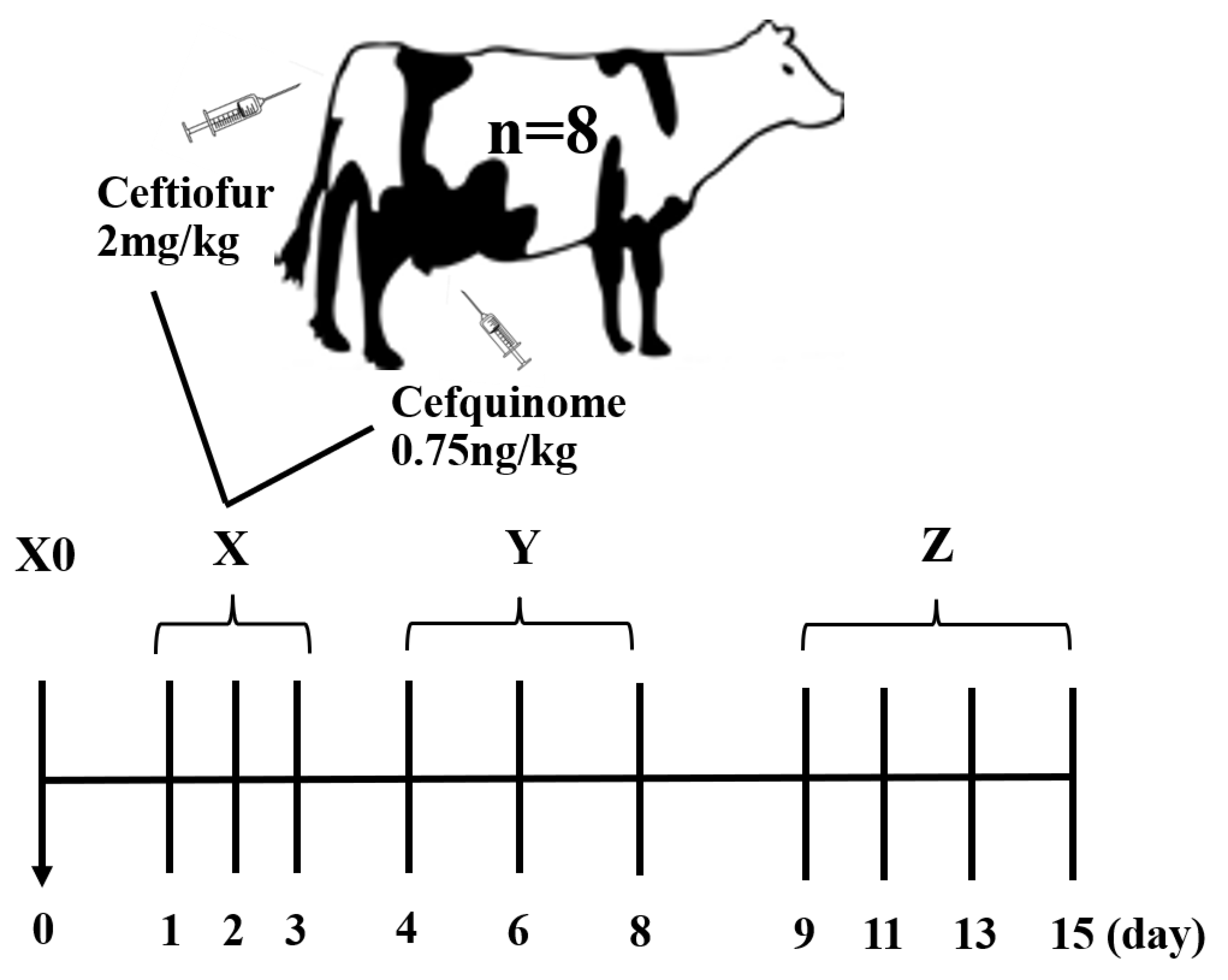
4.1. Animals and Sample Collections
4.2. DNA Extraction
4.3. PCR Amplification
4.4. Illumina Novaseq 6000 Sequencing
4.5. Quantification of Antibiotic Resistance Genes
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Halasa, T.; Huijps, K.; Østerås, O.; Hogeveen, H. Economic effects of bovine mastitis and mastitis management: A review. Vet. Quart. 2007, 29, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.E.; Cerri, R.L.; Ballou, M.A.; Higginbotham, G.E.; Kirk, J.H. Effect of timing of first clinical mastitis occurrence on lactational and reproductive performance of holstein dairy cows. Anim. Reprod. Sci. 2004, 80, 31–45. [Google Scholar] [CrossRef]
- Turk, R.; Piras, C.; Kovai, M.; Samardžija, M.; Ahmed, H.; Canio, M.D.; Urbani, A.; Meštrić, Z.F.; Soggiu, A.; Bonizzi, L.; et al. Proteomics of inflammatory and oxidative stress response in cows with subclinical and clinical mastitis. J. Proteom. 2012, 75, 4412–4428. [Google Scholar] [CrossRef] [PubMed]
- Huijps, K.; Lam, T.J.; Hogeveen, H. Costs of mastitis: Facts and perception. J. Dairy Res. 2008, 75, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Kuipers, A.; Koops, W.J.; Wemmenhove, H. Antibiotic use in dairy herds in the Netherlands from 2005 to 2012. J. Dairy Sci. 2016, 99, 1632–1648. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Wang, Y.; Sun, Y.; Ma, L.; Zeng, Q.; Jiang, X.; Li, A.; Zeng, Z.; Zhang, T. Antibiotic-mediated changes in the fecal microbiome of broiler chickens define the incidence of antibiotic resistance genes. Microbiome 2018, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Holman, D.B.; Yang, W.; Alexander, T.W. Antibiotic treatment in feedlot cattle: A longitudinal study of the effect of oxytetracycline and tulathromycin on the fecal and nasopharyngeal microbiota. Microbiome 2019, 7, 86. [Google Scholar] [CrossRef] [Green Version]
- Kaniyamattam, K.; Vries, A.D.; Tauer, L.W.; Grhn, Y.T. Economics of reducing antibiotic usage for clinical mastitis and metritis through genomic selection-sciencedirect. J. Dairy Sci. 2020, 103, 473–491. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson-Palme, J.; Larsson, D.G.J. Antibiotic resistance genes in the environment: Prioritizing risks. Nat. Rev. Microbiol. 2015, 13, 396. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Wang, H.; Cai, L.; Yu, Y. Prevalence of antibiotic resistance genes and bacterial pathogens in long-term manured greenhouse soils as revealed by metagenomic survey. Environ. Sci. Technol. 2015, 49, 1095–1104. [Google Scholar] [CrossRef]
- Chambers, L.; Yang, Y.; Littier, H.; Ray, P.; Zhang, T.; Pruden, A. Metagenomic analysis of antibiotic resistance genes in dairy cow feces following therapeutic administration of third generation cephalosporin. PLoS ONE 2015, 10, e0133764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, D.M.; Jacob, M.E.; Farmer, K.A.; Callahan, B.J.; Papich, M.G. Ceftiofur formulation differentially affects the intestinal drug concentration, resistance of fecal Escherichia coli, and the microbiome of steers. PLoS ONE 2019, 14, e0223378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- USDA. Dairy 2014. Milk Quality, Milking Procedures, and Mastitis in the United States; USDA–APHIS–VS–CEAH–NAHMS; USDA: Washington, DC, USA, 2016.
- Wang, J.; Shan, Q.; Ding, H.; Liang, C.; Zeng, Z. Pharmacodynamics of cefquinome in a neutropenic mouse thigh model of Staphylococcus aureus infection. Antimicrob. Agents Chemother. 2014, 58, 3008–3012. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhao, D.; Yu, Y.; Yang, X.; Shi, W.; Peng, Y. Pharmacokinetics, bioavailability, and PK/PD relationship of cefquinome for Escherichia coli in beagle dogs. J. Vet. Pharmacol. Ther. 2015, 38, 543–548. [Google Scholar] [CrossRef]
- Guo, C.; Liao, X.; Wang, M.; Wang, F.; Yan, C. In vivo pharmacodynamics of Cefquinome in a neutropenic mouse thigh model of Streptococcus suis serotype 2 at varied initial inoculum sizes. Antimicrob. Agents Chemother. 2016, 60, 1114–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Feng, B.; Gu, X.; Yang, D.; Zeng, Z.; Zhang, B.; Ding, H. Correlation of PK/PD Indices with Resistance Selection for Cefquinome against Staphylococcus aureus in an In Vitro Model. Front. Microbiol. 2016, 7, 466. [Google Scholar] [CrossRef]
- Pehlivanoglu, F.; Turutoglu, H.; Ozturk, D. CTX-M-15-Type Extended-Spectrum Beta-Lactamase-Producing Escherichia coli as Causative Agent of Bovine Mastitis. Foodborne Pathog. Dis. 2016, 13, 477–482. [Google Scholar] [CrossRef]
- Stannarius, C.; Bürgi, E.; Regula, G.; Zychowska, M.A.; Stephan, R. Antimicrobial resistance in Escherichia coli strains isolated from swiss weaned pigs and sows. Schweiz Arch Tierheilkd 2009, 151, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.H.; Wei, S.Y.; Ma, J.Y.; Zeng, Z.L.; Lü, D.H.; Yang, G.X.; Chen, Z.L. Detection and characterization of CTX-M and CMY-2 b-lactamases among Escherichia coli isolates from farm animals in Guangdong province of China. Int. J. Antimicrob. 2007, 29, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; White, D.G.; Mcdermott, P.F.; Friedman, S.; English, L.; Ayers, S.; Ayers, S.; Meng, J.; Maurer, J.J.; Holland, R.; et al. Identification and expression of cephamycinase blacmy genes in Escherichia coli and Salmonella isolates from food animals and ground meat. Antimicrob. Agents Chemother. 2001, 45, 3647–3650. [Google Scholar] [CrossRef] [Green Version]
- Venturini, C.; Bowring, B.; Fajardo-Lubian, A.; Devine, C.; Iredell, J. Effects of antibiotic treatment with piperacillin/tazobactam versus ceftriaxone on the composition of the murine gut microbiota. Antimicrob. Agents Chemother. 2020, 65, 65. [Google Scholar] [CrossRef] [PubMed]
- Dorado-García, A.; Mevius, D.J.; Jacobs, J.J.; Van Geijlswijk, I.M.; Mouton, J.W.; Wagenaar, J.A.; Heederik, D.J. Quantitative assessment of antimicrobial resistance in livestock during the course of a nationwide antimicrobial use reduction in the Netherlands. J. Antimicrob. Chemother. 2016, 71, 3607–3619. [Google Scholar] [CrossRef] [Green Version]
- Chander, Y.; Oliveira, S.; Goyal, S.M. Characterisation of ceftiofur resistance in swine bacterial pathogens. Vet. J. 2011, 187, 139–141. [Google Scholar] [CrossRef]
- Dong, L.; Meng, L.; Liu, H.M.; Wu, H.M.; Hu, H.Y.; Zheng, N.; Wang, J.Q.; Schroyen, M. Effect of therapeutic administration of β-lactam antibiotics on the bacterial community and antibiotic resistance patterns in milk. J. Dairy Sci. 2021, 104, 7018–7025. [Google Scholar] [CrossRef]
- Gāliņa, D.; Balins, A.; Valdovska, A. The Prevalence and Characterization of Fecal Extended-Spectrum-Beta-Lactamase-Producing Escherichia coli Isolated from Pigs on Farms of Different Sizes in Latvia. Antibiotics 2021, 10, 1099. [Google Scholar] [CrossRef]
- García, N.; Gutiérrez, G.; Lorenzo, M.; García, J.E.; Píriz, S.; Quesada, A. Genetic determinants for cfxA expression in Bacteroides strains isolated from human infections. J. Antimicrob. Chemoth. 2008, 62, 942–947. [Google Scholar] [CrossRef] [Green Version]
- Avelar, K.E.; Otsuki, K.; Vicente, A.C.; Vieira, J.M.; de Paula, G.R.; Domingues, R.M.; Ferreira, M.C. Presence of the cfxA gene in Bacteroides distasonis. Res. Microbiol. 2003, 154, 369–374. [Google Scholar] [CrossRef]
- Fernández-Varón, E.; Cárceles-García, C.; Serrano-Rodríguez, J.M.; Cárceles-Rodríguez, C.M. Pharmacokinetics (PK), pharmacodynamics (PD), and PK-PD integration of ceftiofur after a single intravenous, subcutaneous and subcutaneous-LA administration in lactating goats. BMC Vet. Res. 2016, 12, 232. [Google Scholar] [CrossRef] [Green Version]
- Lowrance, T.C.; Loneragan, G.H.; Kunze, D.J.; Platt, T.M.; Ives, S.E.; Scott, H.M.; Norby, B.; Vet, C.M.; Echeverry, A.; Brashears, M.M. Changes in antimicrobial susceptibility in a population of Escherichia coli isolated from feedlot cattle administered ceftiofur crystalline-free acid. Am. J. Vet. Res. 2007, 68, 501. [Google Scholar] [CrossRef]
- Enne, V.I.; Bennett, P.M.; Livermore, D.M.; Hall, L.M. Enhancement of host fitness by the sul2-coding plasmid p9123 in the absence of selective pressure. J. Antimicrob. Chemother. 2004, 53, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Brenciani, A.; Bacciaglia, A.; Vecchi, M.; Vitali, L.A.; Varaldo, P.E.; Giovanetti, E. Genetic elements carrying erm (B) in Streptococcus pyogenes and association with tet (M) tetracycline resistance gene. Antimicrob. Agents Chemother. 2007, 51, 1209–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauch, A.; Götker, S.; Pühler, A.; Kalinowski, J.; Thierbach, G. The 27.8-kb R-plasmid pTET3 from Corynebacterium glutamicum encodes the aminoglycoside adenyltransferase gene cassette aadA9 and the regulated tetracycline efflux system tet33 flanked by active copies of the widespread insertion sequence IS6100. Plasmid 2002, 48, 117–129. [Google Scholar] [CrossRef]
- Su, J.Q.; Wei, B.; Ou-Yang, W.Y.; Huang, F.Y.; Zhao, Y.; Xu, H.J. Antibiotic resistome and its association with bacterial communities during sewage sludge composting. Environ. Sci. Technol. 2015, 49, 7356–7363. [Google Scholar] [CrossRef]
- Huang, X.; Zheng, J.; Tian, S.; Liu, C.; Liu, L.; Wei, L.; Fan, H.; Zhang, T.; Wang, L.; Zhu, G.; et al. Higher temperatures do not always achieve better antibiotic resistance gene removal in anaerobic digestion of swine manure. Appl. Environ. Microbiol. 2019, 85, e02878-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.J.; Stombaugh, K.; Bittinger, F.D.; Bushman, E.K.; Costello, N.; Fierer, A.G.; Peña, J.K.; Goodrich, J.I.; Gordon, G.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.; Ryabin, T. Introducing mothur: Open-Source, PlatformIndependent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Vegan: Community Ecology Package, R Package Version 1.17-4. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 31 December 2021).
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, L.; Meng, L.; Liu, H.; Wu, H.; Schroyen, M.; Zheng, N.; Wang, J. Effect of Cephalosporin Treatment on the Microbiota and Antibiotic Resistance Genes in Feces of Dairy Cows with Clinical Mastitis. Antibiotics 2022, 11, 117. https://doi.org/10.3390/antibiotics11010117
Dong L, Meng L, Liu H, Wu H, Schroyen M, Zheng N, Wang J. Effect of Cephalosporin Treatment on the Microbiota and Antibiotic Resistance Genes in Feces of Dairy Cows with Clinical Mastitis. Antibiotics. 2022; 11(1):117. https://doi.org/10.3390/antibiotics11010117
Chicago/Turabian StyleDong, Lei, Lu Meng, Huimin Liu, Haoming Wu, Martine Schroyen, Nan Zheng, and Jiaqi Wang. 2022. "Effect of Cephalosporin Treatment on the Microbiota and Antibiotic Resistance Genes in Feces of Dairy Cows with Clinical Mastitis" Antibiotics 11, no. 1: 117. https://doi.org/10.3390/antibiotics11010117