
Kinase Inhibitor Library Screening Identifies the Cancer Therapeutic Sorafenib and Structurally Similar Compounds as Strong Inhibitors of the Fungal Pathogen Histoplasma capsulatum

 ,
,
Abstract
:1. Introduction
2. Results
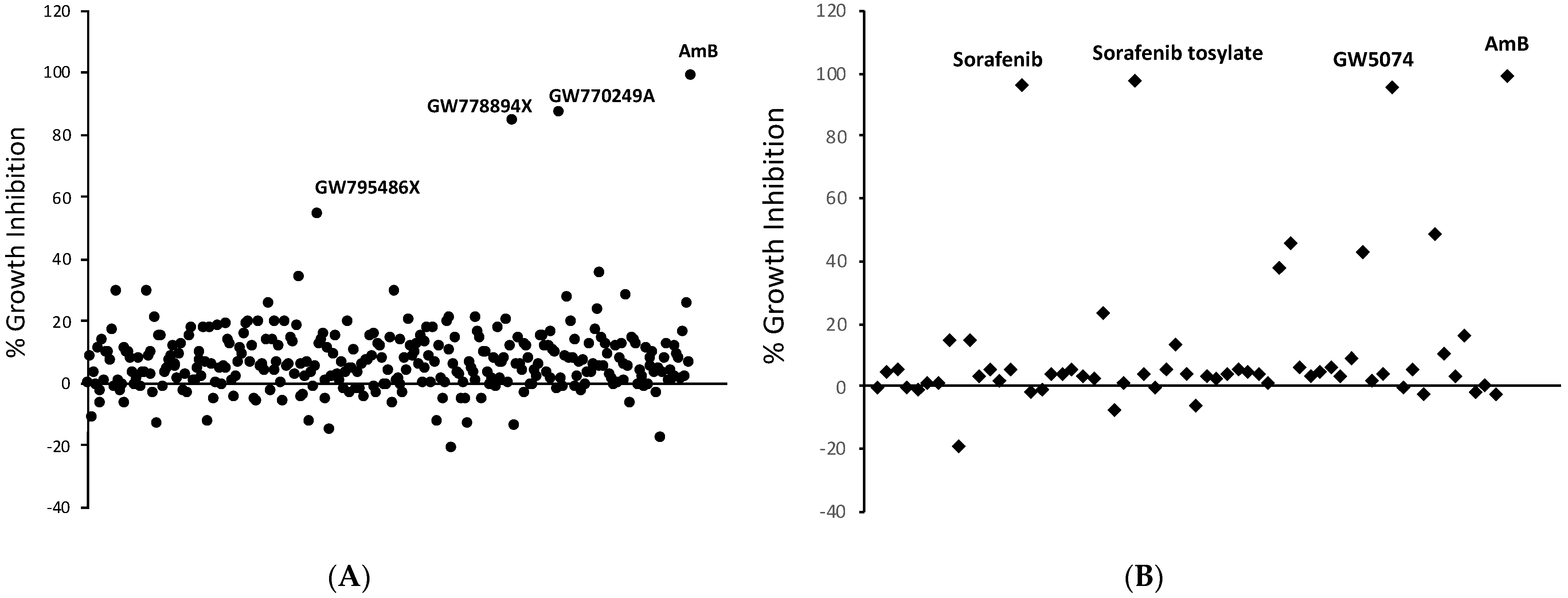
2.1. Kinase Inhibitor Library Screening Identifies Anti-Histoplasma Compounds
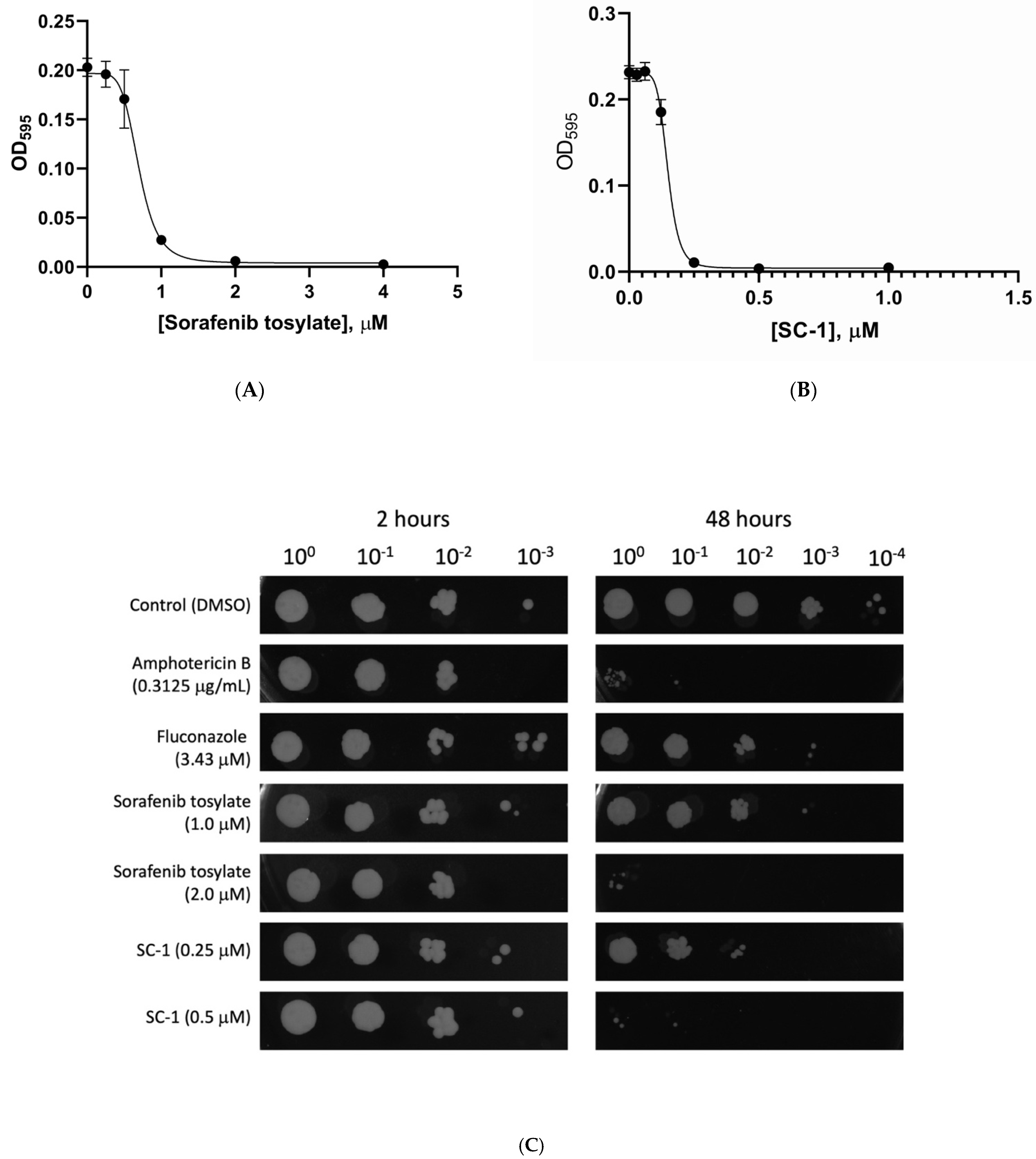
2.2. Characterization of Sorafenib and SC−1 Antifungal Properties
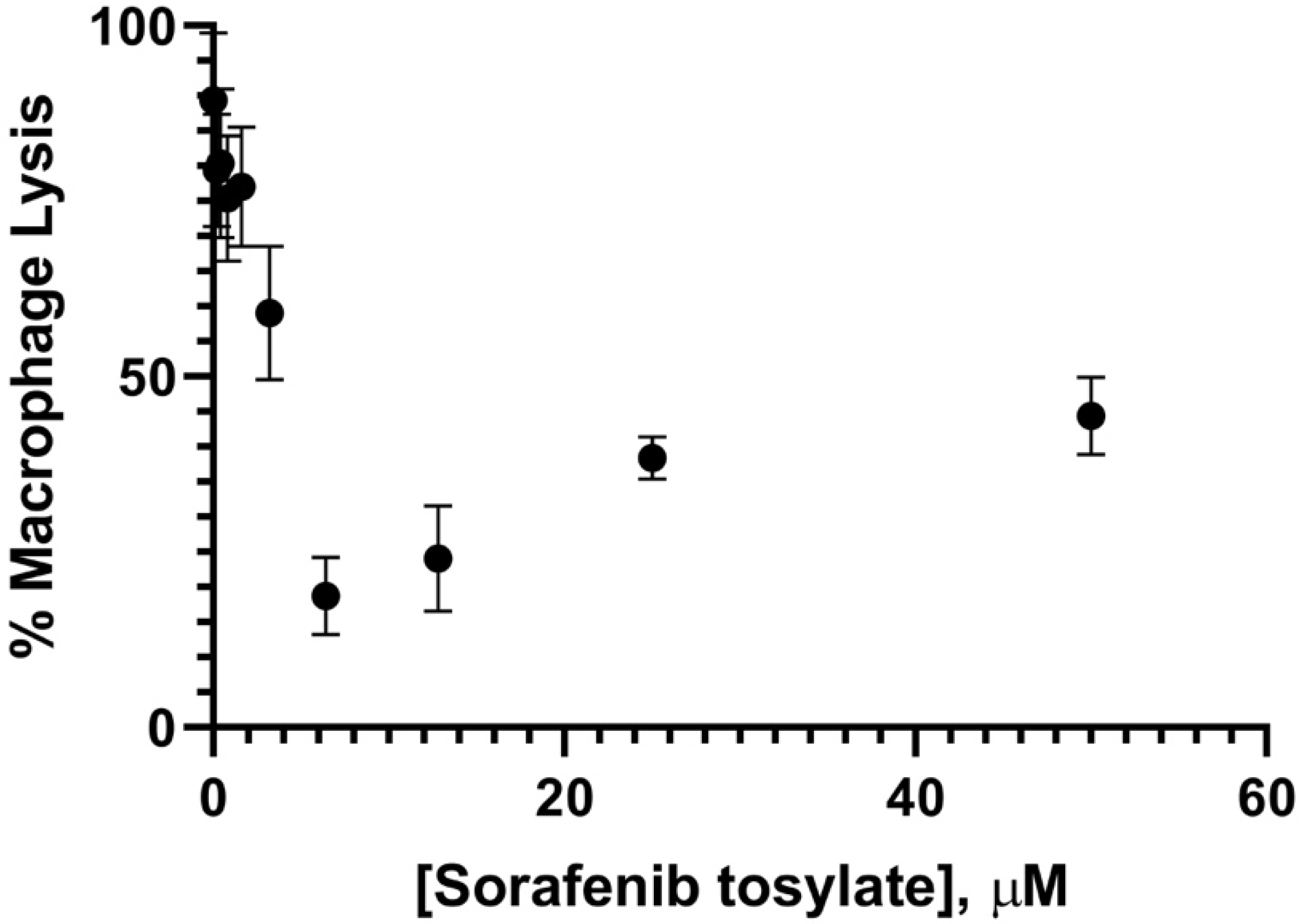
2.3. Effectiveness of Sorafenib and SC−1 against Intracellular H. capsulatum
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Fungal Strains and Culture
5.2. Chemical Library Screening
5.3. MIC Determination
5.4. LogP Determination
5.5. Fungistatic/Fungicidal Testing
5.6. Macrophage Lysis Assay
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pfaller, M.A.; Diekema, D. Epidemiology of Invasive Mycoses in North America. Crit. Rev. Microbiol. 2010, 36, 1–53. [Google Scholar] [CrossRef] [PubMed]
- Benedict, K.; Jackson, B.R.; Chiller, T.; Beer, K.D. Estimation of Direct Healthcare Costs of Fungal Diseases in the United States. Clin. Infect. Dis. 2019, 68, 1791–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwath, M.C.; Fecher, R.A.; Deepe, G.S. Histoplasma capsulatum, lung infection and immunity. Futur. Microbiol. 2015, 10, 967–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, V.V.; Evans, T.; Peterson, M.W. Reactivation histoplasmosis after treatment with anti-tumor necrosis factor α in a patient from a nonendemic area. Respir. Med. 2006, 100, 1291–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergidis, P.; Avery, R.K.; Wheat, L.J.; Dotson, J.L.; Assi, M.A.; Antoun, S.A.; Hamoud, K.A.; Burdette, S.D.; Freifeld, A.G.; McKinsey, D.S.; et al. Histoplasmosis Complicating Tumor Necrosis Factor–α Blocker Therapy: A Retrospective Analysis of 98 Cases. Clin. Infect. Dis. 2015, 61, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Nakelchik, M.; Mangino, J.E. Reactivation of histoplasmosis after treatment with infliximab. Am. J. Med. 2002, 112, 78–79. [Google Scholar] [CrossRef]
- Lucey, O.; Carroll, I.; Bjorn, T.; Millar, M. Reactivation of latent Histoplasma and disseminated cytomegalovirus in a returning traveller with ulcerative colitis. JMM Case Rep. 2018, 5, e005170. [Google Scholar] [CrossRef]
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017, 16, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Odds, F.C.; Brown, A.J.; Gow, N.A. Antifungal agents: Mechanisms of action. Trends Microbiol. 2003, 11, 272–279. [Google Scholar] [CrossRef]
- Wheat, L.J.; Freifeld, A.G.; Kleiman, M.B.; Baddley, J.W.; McKinsey, D.S.; Loyd, J.; Kauffman, C.A. Clinical Practice Guidelines for the Management of Patients with Histoplasmosis: 2007 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2007, 45, 807–825. [Google Scholar] [CrossRef] [Green Version]
- Nett, J.E.; Andes, D.R. Antifungal Agents: Spectrum of Activity, Pharmacology, and Clinical Indications. Infect. Dis. Clin. North Am. 2016, 30, 51–83. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.W.; Su, L.; Yu, D.T.; Chertow, G.M.; Seger, D.L.; Gomes, D.R.J.; Dasbach, E.J.; Platt, R. Mortality and Costs of Acute Renal Failure Associated with Amphotericin B Therapy. Clin. Infect. Dis. 2001, 32, 686–693. [Google Scholar] [CrossRef]
- Pappas, P.G.; Kauffman, C.A.; Andes, D.; Clancy, C.J.; Marr, K.A.; Ostrosky-Zeichner, L.; Reboli, A.C.; Schuster, M.G.; Vazquez, J.A.; Walsh, T.J.; et al. Clinical Practice Guideline for the Management of Candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2016, 62, e1–e50. [Google Scholar] [CrossRef]
- Nakai, T.; Uno, J.; Ikeda, F.; Tawara, S.; Nishimura, K.; Miyaji, M. In Vitro Antifungal Activity of Micafungin (FK463) against Dimorphic Fungi: Comparison of Yeast-Like and Mycelial Forms. Antimicrob. Agents Chemother. 2003, 47, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Hage, C.A.; Connolly, P.; Horan, D.; Durkin, M.; Smedema, M.; Zarnowski, R.; Smith, P.; Wheat, L.J. Investigation of the Efficacy of Micafungin in the Treatment of Histoplasmosis Using Two North American Strains of Histoplasma capsulatum. Antimicrob. Agents Chemother. 2011, 55, 4447–4450. [Google Scholar] [CrossRef] [Green Version]
- Roemer, T.; Krysan, D.J. Antifungal Drug Development: Challenges, Unmet Clinical Needs, and New Approaches. Cold Spring Harb. Perspect. Med. 2014, 4, a019703. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Bamborough, P. System-based drug discovery within the human kinome. Expert Opin. Drug Discov. 2012, 7, 1–18. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A.M.; Bountra, C.; Kerr, D.J.; Willson, T.M. Open access chemical and clinical probes to support drug discovery. Nat. Chem. Biol. 2009, 5, 436–440. [Google Scholar] [CrossRef]
- Hohmann, S. Osmotic Stress Signaling and Osmoadaptation in Yeasts. Microbiol. Mol. Biol. Rev. 2002, 66, 300–372. [Google Scholar] [CrossRef] [Green Version]
- Andrews, P.; Stark, M. Type 1 protein phosphatase is required for maintenance of cell wall integrity, morphogenesis and cell cycle progression in Saccharomyces cerevisiae. J. Cell Sci. 2000, 113, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Gustin, M.C.; Albertyn, J.; Alexander, M.; Davenport, K. MAP Kinase Pathways in the Yeast Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1998, 62, 1264–1300. [Google Scholar] [CrossRef] [Green Version]
- Posas, F. Osmotic Activation of the HOG MAPK Pathway via Ste11p MAPKKK: Scaffold Role of Pbs2p MAPKK. Science 1997, 276, 1702–1705. [Google Scholar] [CrossRef] [PubMed]
- Wojda, I.; Monge, R.A.; Bebelman, J.-P.; Mager, W.H.; Siderius, M. Response to high osmotic conditions and elevated temperature in Saccharomyces cerevisiae is controlled by intracellular glycerol and involves coordinate activity of MAP kinase pathways. Microbiology 2003, 149, 1193–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leberer, E.; Harcus, D.; Dignard, D.; Johnson, L.; Ushinsky, S.; Thomas, D.; Schröppel, K. Ras links cellular morphogenesis to virulence by regulation of the MAP kinase and cAMP signalling pathways in the pathogenic fungus Candida albicans. Mol. Microbiol. 2008, 42, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Csank, C.; Makris, C.; Meloche, S.; Schröppel, K.; Röllinghoff, M.; Dignard, D.; Thomas, D.; Whiteway, M. Derepressed Hyphal Growth and Reduced Virulence in a VH1 Family-related Protein Phosphatase Mutant of the Human Pathogen Candida albicans. Mol. Biol. Cell 1997, 8, 2539–2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Du, W.; Zhao, J.; Zhang, L.; Zhu, Z.; Jiang, L. The MAP kinase-activated protein kinase Rck2p regulates cellular responses to cell wall stresses, filamentation and virulence in the human fungal pathogen Candida albicans. FEMS Yeast Res. 2010, 10, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-T.; So, Y.-S.; Yang, D.-H.; Jung, K.-W.; Choi, J.; Lee, D.-G.; Kwon, H.; Jang, J.; Wang, L.L.; Cha, S.; et al. Systematic functional analysis of kinases in the fungal pathogen Cryptococcus neoformans. Nat. Commun. 2016, 7, 12766. [Google Scholar] [CrossRef]
- Bahn, Y.-S.; Kojima, K.; Cox, G.M.; Heitman, J. Specialization of the HOG Pathway and Its Impact on Differentiation and Virulence of Cryptococcus neoformans. Mol. Biol. Cell 2005, 16, 2285–2300. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-Y.; Ko, Y.-J.; Jung, K.-W.; Strain, A.; Nielsen, K.; Bahn, Y.-S. Hrk1 Plays Both Hog1-Dependent and -Independent Roles in Controlling Stress Response and Antifungal Drug Resistance in Cryptococcus neoformans. PLoS ONE 2011, 6, e18769. [Google Scholar] [CrossRef]
- Manning, G.; Plowman, G.D.; Hunter, T.; Sudarsanam, S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002, 27, 514–520. [Google Scholar] [CrossRef]
- Kojima, K.; Bahn, Y.-S.; Heitman, J. Calcineurin, Mpk1 and Hog1 MAPK pathways independently control fludioxonil antifungal sensitivity in Cryptococcus neoformans. Microbiology 2006, 152, 591–604. [Google Scholar] [CrossRef] [Green Version]
- Elkins, J.M.; Fedele, V.; Szklarz, M.; Azeez, K.R.A.; Salah, E.; Mikolajczyk, J.; Romanov, S.; Sepetov, N.; Huang, X.-P.; Roth, B.L.; et al. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol. 2016, 34, 95–103. [Google Scholar] [CrossRef]
- Drewry, D.; Willson, T.M.; Zuercher, W.J. Seeding Collaborations to Advance Kinase Science with the GSK Published Kinase Inhibitor Set (PKIS). Curr. Top. Med. Chem. 2014, 14, 340–342. [Google Scholar] [CrossRef]
- Edwards, J.A.; Kemski, M.M.; Rappleye, C.A. Identification of an Aminothiazole with Antifungal Activity against Intracellular Histoplasma capsulatum. Antimicrob. Agents Chemother. 2013, 57, 4349–4359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goughenour, K.D.; Balada-Llasat, J.-M.; Rappleye, C.A. Quantitative Microplate-Based Growth Assay for Determination of Antifungal Susceptibility of Histoplasma capsulatum Yeasts. J. Clin. Microbiol. 2015, 53, 3286–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, R.B.; Holladay, M.W. The Organic Chemistry of Drug Design and Drug Action. In The Organic Chemistry of Drug Design and Drug Action, 3rd ed.; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.-T.; Cheng, A.-L.; Shiau, C.-W.; Huang, H.-P.; Huang, J.-W.; Chen, P.-J.; Chen, K.-F. Signal transducer and activator of transcription 3 is a major kinase-independent target of sorafenib in hepatocellular carcinoma. J. Hepatol. 2011, 55, 1041–1048. [Google Scholar] [CrossRef]
- Chiu, C.-M.; Huang, S.-Y.; Chang, S.-F.; Liao, K.-F.; Chiu, S.-C. Synergistic antitumor effects of tanshinone IIA and sorafenib or its derivative SC-1 in hepatocellular carcinoma cells. OncoTargets Ther. 2018, 11, 1777–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.-H.; Shiau, C.-W.; Jao, P.; Liu, C.-H.; Liu, C.-J.; Tai, W.-T.; Jeng, Y.-M.; Yang, H.-C.; Tseng, T.-C.; Huang, H.-P.; et al. Sorafenib and its derivative SC-1 exhibit antifibrotic effects through signal transducer and activator of transcription 3 inhibition. Proc. Natl. Acad. Sci. USA 2015, 112, 7243–7248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaac, D.T.; Berkes, C.A.; English, B.C.; Murray, D.H.; Lee, Y.N.; Coady, A.; Sil, A. Macrophage cell death and transcriptional response are actively triggered by the fungal virulence factor Cbp1 during H.capsulatum infection. Mol. Microbiol. 2015, 98, 910–929. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.; Roncal, N.E.; Lee, P.J.; Leed, S.E.; Erath, J.; Rodriguez, A.; Sciotti, R.; Pollastri, M.P. Repurposing human Aurora kinase inhibitors as leads for anti-protozoan drug discovery. MedChemComm 2014, 5, 655–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dichiara, M.; Marrazzo, A.; Prezzavento, O.; Collina, S.; Rescifina, A.; Amata, E. Repurposing of Human Kinase Inhibitors in Neglected Protozoan Diseases. ChemMedChem 2017, 12, 1235–1253. [Google Scholar] [CrossRef] [Green Version]
- Stylianou, M.; Kulesskiy, E.; Lopes, J.P.; Granlund, M.; Wennerberg, K.; Urban, C.F. Antifungal Application of Nonantifungal Drugs. Antimicrob. Agents Chemother. 2013, 58, 1055–1062. [Google Scholar] [CrossRef] [Green Version]
- Butts, A.; DiDone, L.; Koselny, K.; Baxter, B.K.; Chabrier-Rosello, Y.; Wellington, M.; Krysan, D.J. A Repurposing Approach Identifies Off-Patent Drugs with Fungicidal Cryptococcal Activity, a Common Structural Chemotype, and Pharmacological Properties Relevant to the Treatment of Cryptococcosis. Eukaryot. Cell 2012, 12, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Khalil, A.; Edwards, J.A.; Rappleye, C.A.; Tjarks, W. Design, synthesis, and biological evaluation of aminothiazole derivatives against the fungal pathogens Histoplasma capsulatum and Cryptococcus neoformans. Bioorganic Med. Chem. 2015, 23, 532–547. [Google Scholar] [CrossRef] [Green Version]
- Catalano, A.; Iacopetta, D.; Sinicropi, M.S.; Franchini, C. Diarylureas as Antitumor Agents. Appl. Sci. 2021, 11, 374. [Google Scholar] [CrossRef]
- Garuti, L.; Roberti, M.; Bottegoni, G.; Ferraro, M. Diaryl Urea: A Privileged Structure in Anticancer Agents. Curr. Med. Chem. 2016, 23, 1528–1548. [Google Scholar] [CrossRef]
- Catalano, A.; Iacopetta, D.; Pellegrino, M.; Aquaro, S.; Franchini, C.; Sinicropi, M. Diarylureas: Repositioning from Antitumor to Antimicrobials or Multi-Target Agents against New Pandemics. Antibiotics 2021, 10, 92. [Google Scholar] [CrossRef]
- Iacopetta, D.; Catalano, A.; Ceramella, J.; Saturnino, C.; Salvagno, L.; Ielo, I.; Drommi, D.; Scali, E.; Plutino, M.; Rosace, G.; et al. The Different Facets of Triclocarban: A Review. Molecules 2021, 26, 2811. [Google Scholar] [CrossRef] [PubMed]
- Catalano, A.; Rosato, A.; Salvagno, L.; Iacopetta, D.; Ceramella, J.; Fracchiolla, G.; Sinicropi, M.; Franchini, C. Benzothiazole-Containing Analogues of Triclocarban with Potent Antibacterial Activity. Antibiotics 2021, 10, 803. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, L.; Brahms, A.; Hooper, I.; Carey, B.D.; Lin, S.-C.; Dahal, B.; Narayanan, A.; Kehn-Hall, K. Repurposed FDA-Approved drug sorafenib reduces replication of Venezuelan equine encephalitis virus and other alphaviruses. Antivir. Res. 2018, 157, 57–67. [Google Scholar] [CrossRef]
- Kocyigit-Kaymakcioglu, B.; Celen, A.O.; Tabanca, N.; Ali, A.; Khan, S.I.; Khan, I.A.; Wedge, D.E. Synthesis and Biological Activity of Substituted Urea and Thiourea Derivatives Containing 1,2,4-Triazole Moieties. Molecules 2013, 18, 3562–3576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Poeta, M.; Chen, S.-F.; Von Hoff, D.; Dykstra, C.C.; Wani, M.C.; Manikumar, G.; Heitman, J.; Wall, M.E.; Perfect, J.R. Comparison of In Vitro Activities of Camptothecin and Nitidine Derivatives against Fungal and Cancer Cells. Antimicrob. Agents Chemother. 1999, 43, 2862–2868. [Google Scholar] [CrossRef] [Green Version]
- Heitman, J.; Movva, N.; Hall, M. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef]
- Cutler, N.S.; Heitman, J.; Cardenas, M.E. STT4 Is an Essential Phosphatidylinositol 4-Kinase That Is a Target of Wortmannin in Saccharomyces cerevisiae. J. Biol. Chem. 1997, 272, 27671–27677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib Blocks the RAF/MEK/ERK Pathway, Inhibits Tumor Angiogenesis, and Induces Tumor Cell Apoptosis in Hepatocellular Carcinoma Model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coriat, R.; Nicco, C.; Chéreau, C.; Mir, O.; Alexandre, J.; Ropert, S.; Weill, B.; Chaussade, S.; Goldwasser, F.; Batteux, F. Sorafenib-Induced Hepatocellular Carcinoma Cell Death Depends on Reactive Oxygen Species Production In Vitro and In Vivo. Mol. Cancer Ther. 2012, 11, 2284–2293. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-Y.; Tseng, L.-M.; Su, J.-C.; Chang, K.-C.; Chu, P.-Y.; Tai, W.-T.; Shiau, C.-W.; Chen, K.-F. Novel sorafenib analogues induce apoptosis through SHP-1 dependent STAT3 inactivation in human breast cancer cells. Breast Cancer Res. 2013, 15, R63. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Mehrabi, R.; Xu, J.-R. Mitogen-Activated Protein Kinase Pathways and Fungal Pathogenesis. Eukaryot. Cell 2007, 6, 1701–1714. [Google Scholar] [CrossRef] [Green Version]
- Knauth, P.; Reichenbach, H. On the Mechanism of Action of the Myxobacterial Fungicide Ambruticin. J. Antibiot. 2000, 53, 1182–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion, C.L.; Rappleye, C.; Engle, J.T.; Goldman, W.E. An α-(1,4)-amylase is essential for α -(1,3)-glucan production and virulence in Histoplasma capsulatum. Mol. Microbiol. 2006, 62, 970–983. [Google Scholar] [CrossRef] [PubMed]
- Isaac, D.T.; Coady, A.; Van Prooyen, N.; Sil, A. The 3-Hydroxy-Methylglutaryl Coenzyme a Lyase HCL1 Is Required for Macrophage Colonization by Human Fungal Pathogen Histoplasma capsulatum. Infect. Immun. 2013, 81, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rex, J.H.; Alexander, B.D.; Andes, D.; Arthington-Skaggs, B.; Brown, S.D.; Chaturvedi, V.; Ghannoum, M.A.; Espinel-Ingroff, A.; Knapp, C.C.; Ostrosky-Zeichner, L.; et al. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: Approved Standard, 3rd ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008. [Google Scholar]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, A.Y.; Wang, R.; Lai, L. Computation of Octanol−Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]





{kind=link}
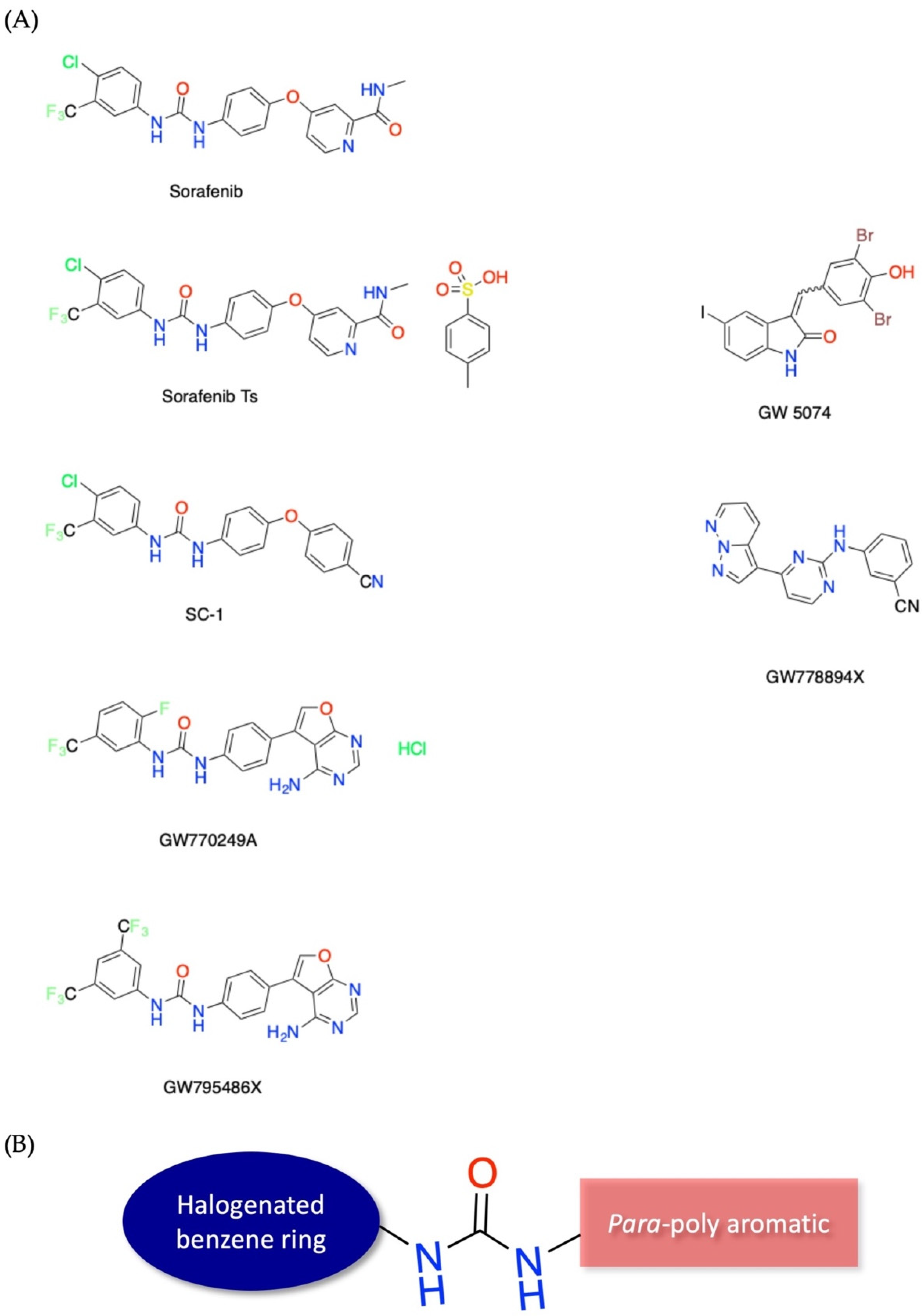{kind=link}
{kind=link}
{kind=link}
| Library | Compound | Hc WU15 | Hc WU8 | C. neoformans | C. albicans | Log P | Satisfies Lipinski’s Rule of 5? |
|---|---|---|---|---|---|---|---|
| GW770249A | 0.5 | 2.0 | >50 | >50 | 3.65 | Yes | |
| PKIS | GW7788994X | 0.25 | 1.0 | >50 | >50 | 1.21 | Yes |
| GW795486X | 2.0 | 4.0 | >50 | >50 | 4.41 | Yes | |
| GW5074 | 2.0 | 4.0 | >50 | >50 | 5.31 | Partially | |
| SelleckChem | Sorafenib tosylate | 1.0 | 2.0 | 25 | >50 | 3.76 | Yes |
| Sorafenib | 1.0 | 2.0 | 25 | >50 | 3.8 | Yes | |
| - | SC-1 | 0.25 | 1.0 | 12.5 | >50 | 5.56 | Partially |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berkes, C.; Franco, J.; Lawson, M.; Brann, K.; Mermelstein, J.; Laverty, D.; Connors, A. Kinase Inhibitor Library Screening Identifies the Cancer Therapeutic Sorafenib and Structurally Similar Compounds as Strong Inhibitors of the Fungal Pathogen Histoplasma capsulatum. Antibiotics 2021, 10, 1223. https://doi.org/10.3390/antibiotics10101223
Berkes C, Franco J, Lawson M, Brann K, Mermelstein J, Laverty D, Connors A. Kinase Inhibitor Library Screening Identifies the Cancer Therapeutic Sorafenib and Structurally Similar Compounds as Strong Inhibitors of the Fungal Pathogen Histoplasma capsulatum. Antibiotics. 2021; 10(10):1223. https://doi.org/10.3390/antibiotics10101223
Chicago/Turabian StyleBerkes, Charlotte, Jimmy Franco, Maxx Lawson, Katelynn Brann, Jessica Mermelstein, Daniel Laverty, and Allison Connors. 2021. "Kinase Inhibitor Library Screening Identifies the Cancer Therapeutic Sorafenib and Structurally Similar Compounds as Strong Inhibitors of the Fungal Pathogen Histoplasma capsulatum" Antibiotics 10, no. 10: 1223. https://doi.org/10.3390/antibiotics10101223