On-Chip Electromembrane Surrounded Solid Phase Microextraction for Determination of Tricyclic Antidepressants from Biological Fluids Using Poly(3,4-ethylenedioxythiophene)—Graphene Oxide Nanocomposite as a Fiber Coating

Abstract
:1. Introduction
2. Experimental
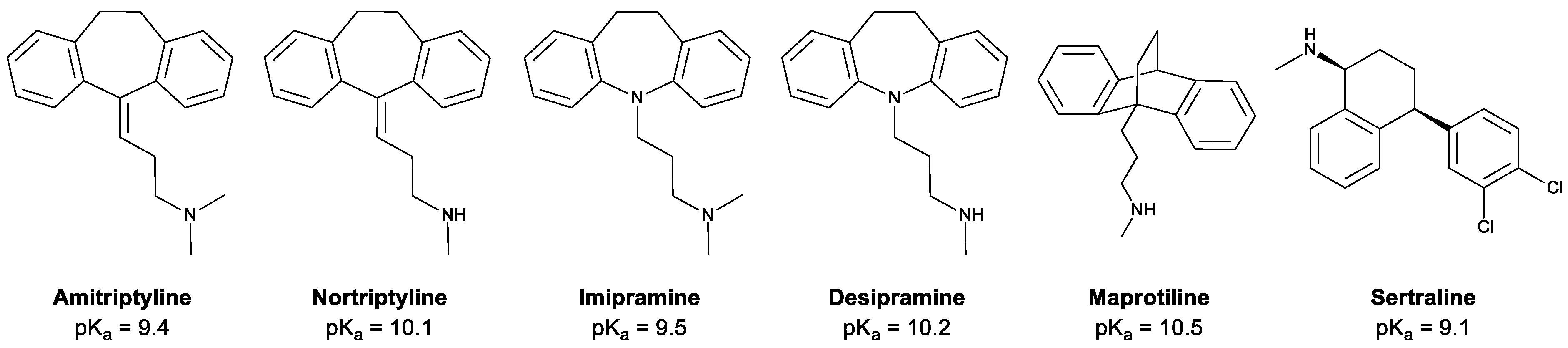
2.1. Chemicals and Materials
2.2. Instruments and Apparatus
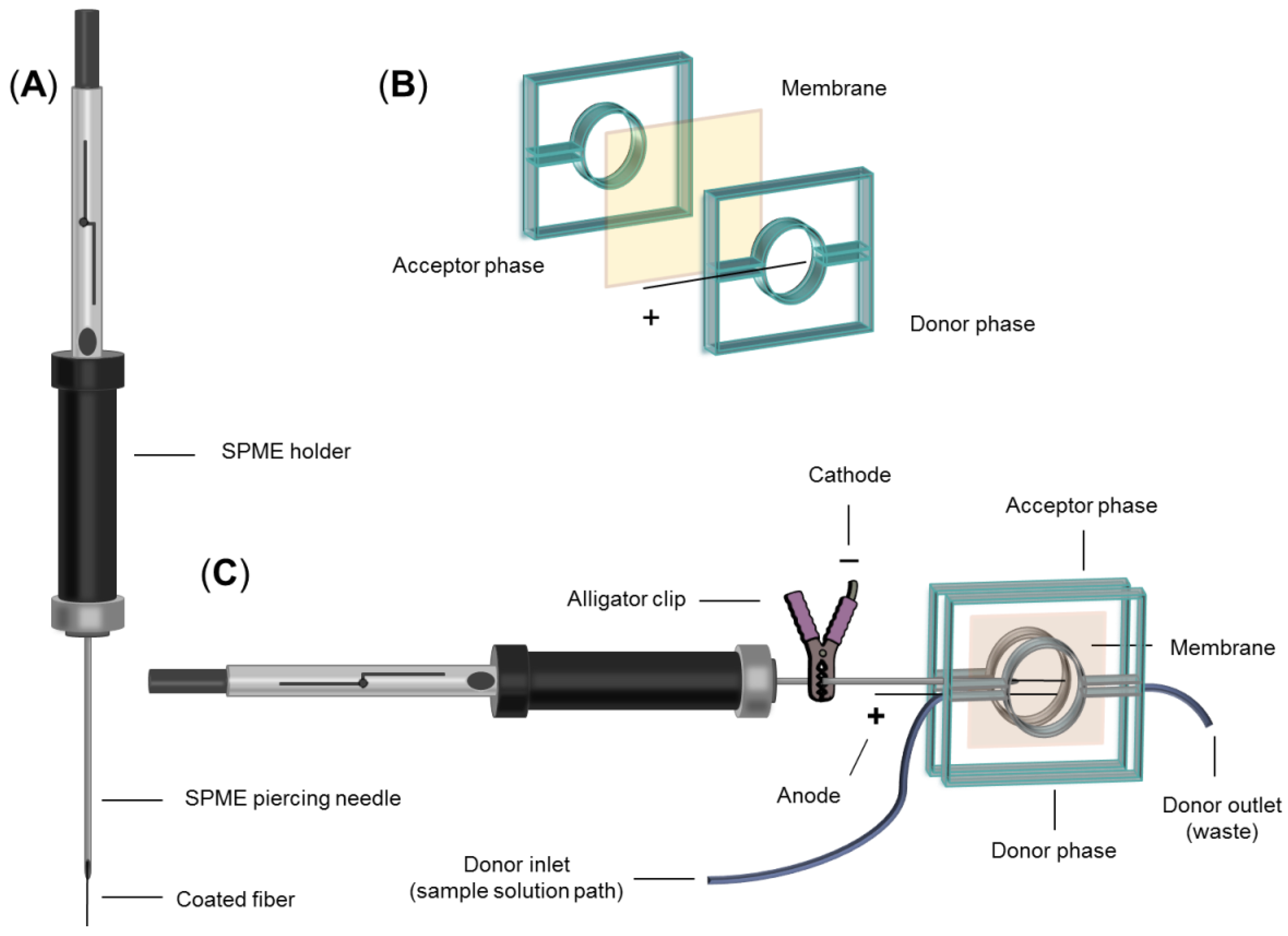
2.3. Fabrication of EM-SPME Chip Device
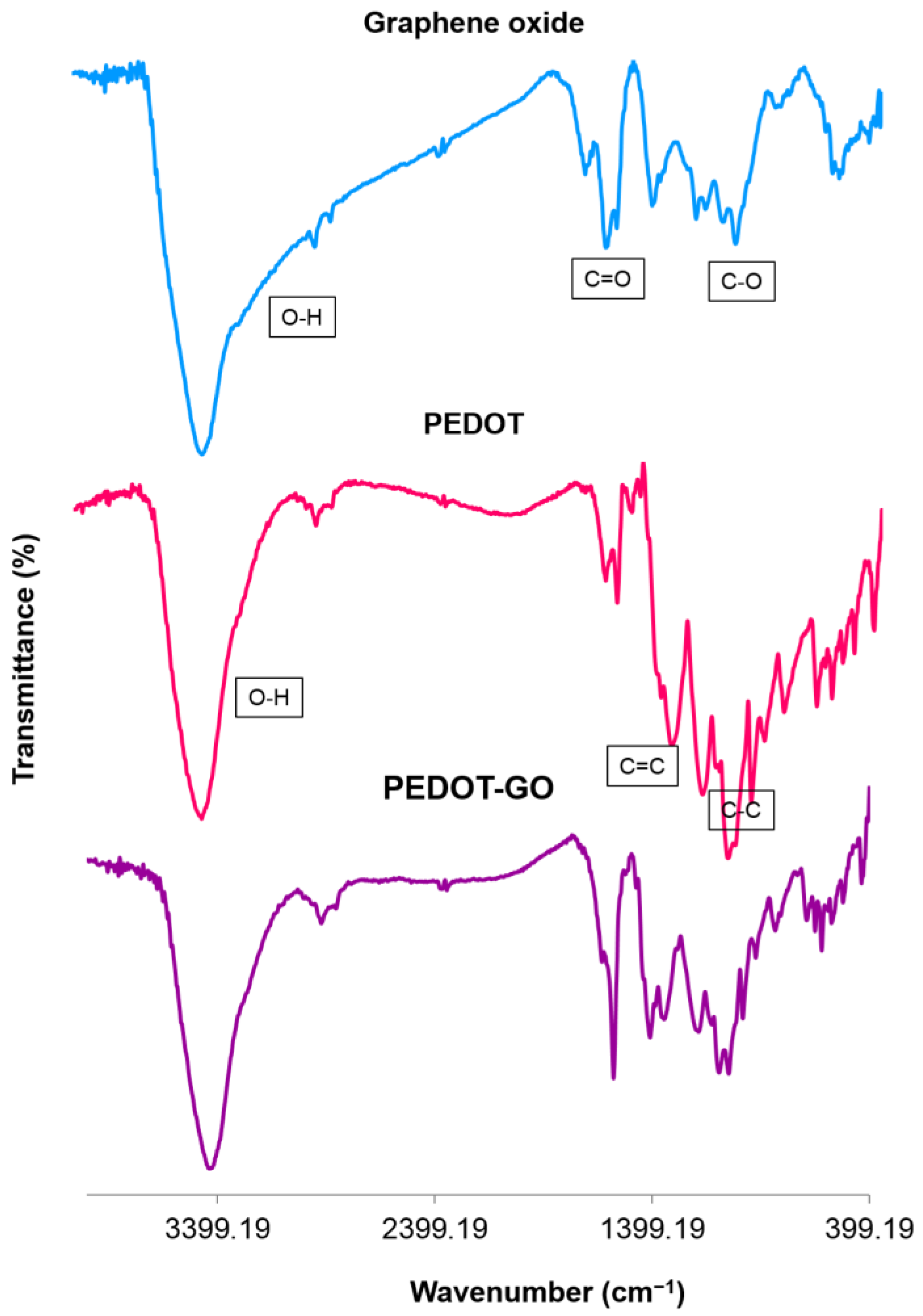
2.4. Synthesis of PEDOT-GO Nanocomposite
2.5. Extraction Procedure
2.6. Real Sample Preparation
3. Results and Discussion
 ). Thus, GO dopant with a negative charge could be incorporated during the reaction into the PEDOT structure to maintain electrical neutrality. Subsequently, the nanocomposite would be electrodeposited on the fiber as a result of electrostatic interaction [33].
). Thus, GO dopant with a negative charge could be incorporated during the reaction into the PEDOT structure to maintain electrical neutrality. Subsequently, the nanocomposite would be electrodeposited on the fiber as a result of electrostatic interaction [33].3.1. Characterization of PEDOT-GO
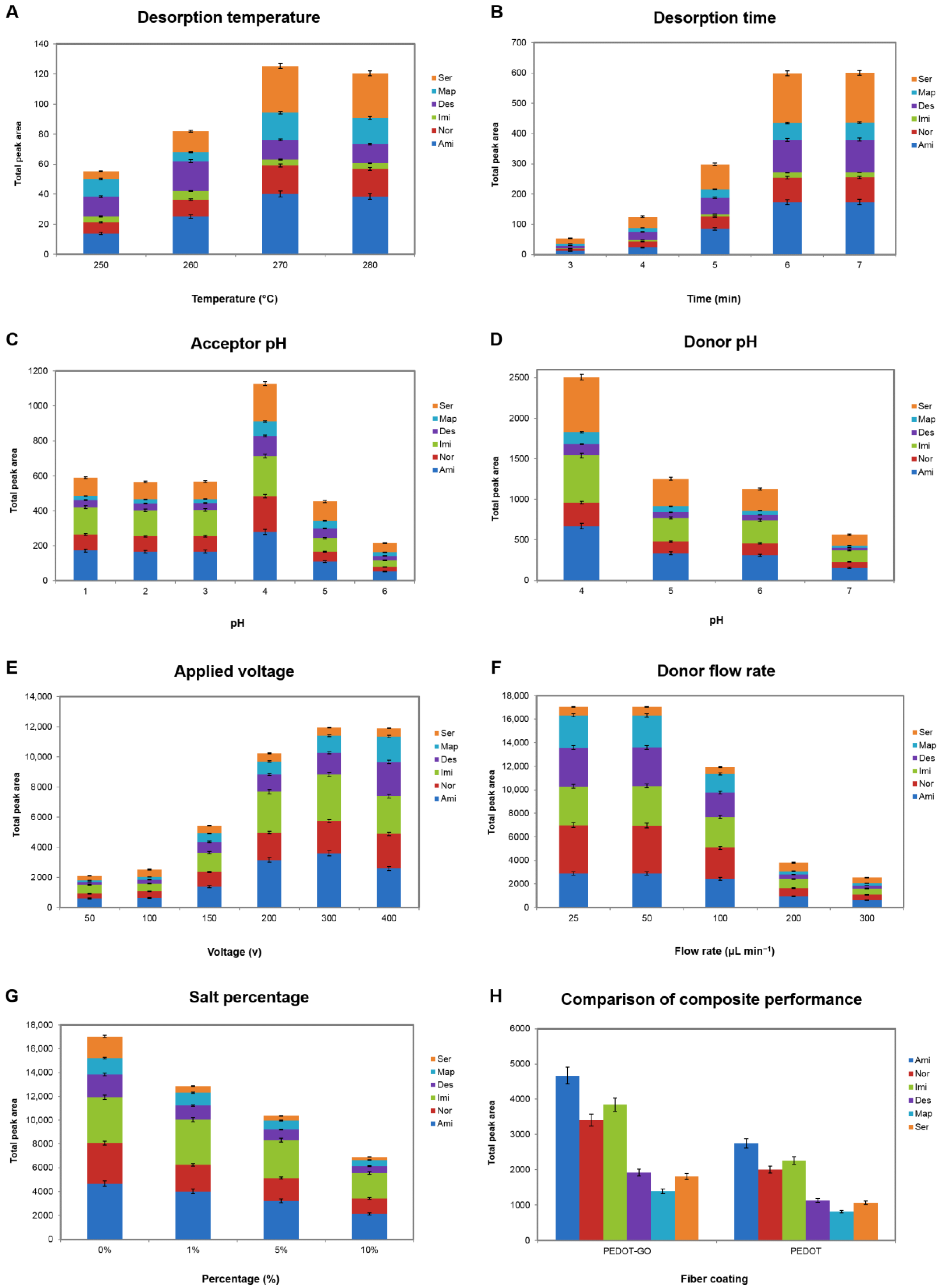
3.2. Optimization of Effective Parameters on the Extraction
3.2.1. Desorption Temperature
3.2.2. Desorption Time
3.2.3. Acceptor pH
3.2.4. Donor pH
3.2.5. Applied Voltage
3.2.6. Sample Solution Flow Rate
3.2.7. Salt Percentage
3.3. Method Validation
3.4. Real Samples Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Poklis, J.L.; Wolf, C.E.; Goldstein, A.; Wolfe, M.L.; Poklis, A. Detection and quantification of tricyclic antidepressants and other psychoactive drugs in urine by HPLC/MS/MS for pain management compliance testing. J. Clin. Lab. Anal. 2012, 26, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Skapinakis, P.; Papatheodorou, T.; Mavreas, V. Antipsychotic augmentation of serotonergic antidepressants in treatment-resistant obsessive–compulsive disorder: A meta-analysis of the randomized controlled trials. Eur. Neuropsychopharmacol. 2007, 17, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Zai, G.; Brandl, E.J.; Müller, D.J.; Richter, M.A.; Kennedy, J.L. Pharmacogenetics of antidepressant treatment in obsessive–compulsive disorder: An update and implications for clinicians. Pharmacogenomics 2014, 15, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Verdu, B.; Decosterd, I.; Buclin, T.; Stiefel, F.; Berney, A. Antidepressants for the treatment of chronic pain. Drugs 2008, 68, 2611–2632. [Google Scholar] [CrossRef] [PubMed]
- Von Wolff, A.; Hölzel, L.P.; Westphal, A.; Härter, M.; Kriston, L. Selective serotonin reuptake inhibitors and tricyclic antidepressants in the acute treatment of chronic depression and dysthymia: A systematic review and meta-analysis. J. Affect. Disord. 2013, 144, 7–15. [Google Scholar] [CrossRef]
- Sundström-Poromaa, I.; Bixo, M.; Björn, I.; Nordh, O. Compliance to antidepressant drug therapy for treatment of premenstrual syndrome. J. Psychosom. Obstet. Gynaecol. 2000, 21, 205–211. [Google Scholar] [CrossRef]
- Burch, R. Antidepressants for preventive treatment of migraine. Curr. Treat. Options Neurol. 2019, 21, 18. [Google Scholar] [CrossRef]
- Banaschewski, T.; Roessner, V.; Dittmann, R.W.; Janardhanan Santosh, P.; Rothenberger, A. Non-stimulant medications in the treatment of ADHD. Eur. Child Adolesc. Psychiatry 2004, 13, 102–116. [Google Scholar] [CrossRef]
- Buhmann, C.B.; Nordentoft, M.; Ekstroem, M.; Carlsson, J.; Mortensen, E.L. The effect of flexible cognitive–behavioural therapy and medical treatment, including antidepressants on post-traumatic stress disorder and depression in traumatised refugees: Pragmatic randomised controlled clinical trial. Br. J. Psychiatry 2016, 208, 252–259. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, A.M.; Rodrigues Schumacher, S.D.O.; de Souza Antunes, A.M. Antidepressants drugs and addiction treatment: A development view and a technological patent landscape of drugs and compositions. J. Addict. Med. Ther. Sci. 2021, 7, 020–025. [Google Scholar] [CrossRef]
- Gabriel, M.; Sharma, V. Antidepressant discontinuation syndrome. Can. Med. Assoc. J. 2017, 189, 747. [Google Scholar] [CrossRef] [Green Version]
- Riediger, C.; Schuster, T.; Barlinn, K.; Maier, S.; Weitz, J.; Siepmann, T. Adverse effects of antidepressants for chronic pain: A systematic review and meta-analysis. Front. Neurol. 2017, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Himmerich, H.; Minkwitz, J.; Kirkby, K.C. Weight gain and metabolic changes during treatment with antipsychotics and antidepressants. Endocr. Metab. Immune. Disord. Drug Targets 2015, 15, 252–260. [Google Scholar] [CrossRef]
- Friedman, R.A. Antidepressants’ black-box warning—10 years later. N. Engl. J. Med. 2014, 371, 1666–1668. [Google Scholar] [CrossRef]
- Clayton, A.H.; Croft, H.A.; Handiwala, L. Antidepressants and sexual dysfunction: Mechanisms and clinical implications. Postgrad. Med. 2014, 126, 91–99. [Google Scholar] [CrossRef]
- Salamat, Q.; Yamini, Y.; Moradi, M.; Farahani, A.; Feizi, N. Extraction of antidepressant drugs in biological samples using alkanol-based nano structured supramolecular solvent microextraction followed by gas chromatography-mass spectrometric analysis. J. Sep. Sci. 2019, 42, 1620–1628. [Google Scholar] [CrossRef]
- Davarani, S.S.H.; Najarian, A.M.; Nojavan, S.; Tabatabaei, M.A. Electromembrane extraction combined with gas chromatography for quantification of tricyclic antidepressants in human body fluids. Anal. Chim. Acta 2012, 725, 51–56. [Google Scholar] [CrossRef]
- Gjelstad, A.; Pedersen-Bjergaard, S. Electromembrane extraction-Three-phase electrophoresis for future preparative applications. Electrophoresis 2014, 35, 2421–2428. [Google Scholar] [CrossRef]
- Zamani, R.; Yamini, Y. On-chip electromembrane extraction with monolithic adsorbent in acceptor phase. In Proceedings of the 10th National Biennial Seminar (Webinar) of Chemistry and Environment, Quchan University of Technology, Quchan, Iran, 1–2 November 2021; p. 1150. [Google Scholar]
- Pedersen-Bjergaard, S.; Rasmussen, K.E. Electrokinetic migration across artificial liquid membranes: New concept for rapid sample preparation of biological fluids. J. Chromatogr. A 2006, 1109, 183–190. [Google Scholar] [CrossRef]
- Huang, C.; Chen, Z.; Gjelstad, A.; Pedersen-Bjergaard, S.; Shen, X. Electromembrane extraction. Trends Anal. Chem. 2017, 95, 47–56. [Google Scholar] [CrossRef]
- Rezazadeh, M.; Yamini, Y.; Seidi, S.; Ebrahimpour, B. Electromembrane surrounded solid phase microextraction: A novel approach for efficient extraction from complicated matrices. J. Chromatogr. A 2013, 1280, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Rezazadeh, M.; Yamini, Y.; Seidi, S. Application of a new nanocarbonaceous sorbent in electromembrane surrounded solid phase microextraction for analysis of amphetamine and methamphetamine in human urine and whole blood. J. Chromatogr. A 2015, 1396, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nezakati, T.; Seifalian, A.; Tan, A.; Seifalian, A.M. Conductive polymers: Opportunities and challenges in biomedical applications. Chem. Rev. 2018, 118, 6766–6843. [Google Scholar] [CrossRef] [PubMed]
- Groenendaal, L.; Jonas, F.; Freitag, D.; Pielartzik, H.; Reynolds, J.R. Poly(3,4-ethylenedioxythiophene) and its derivatives: Past, present, and future. Adv. Mater. 2000, 12, 481–494. [Google Scholar] [CrossRef]
- Ríos, Á.; Zougagh, M. Sample preparation for micro total analytical systems (μ-TASs). Trends Anal. Chem. 2013, 43, 174–188. [Google Scholar] [CrossRef]
- Ríos, Á.; Zougagh, M.; Avila, M. Miniaturization through lab-on-a-chip: Utopia or reality for routine laboratories? A review. Anal. Chim. Acta 2012, 740, 1–11. [Google Scholar] [CrossRef]
- Alidoust, M.; Yamini, Y.; Baharfar, M.; Seidi, S.; Rasouli, F. Microfluidic-enabled versatile hyphenation of electromembrane extraction and thin film solid phase microextraction. Talanta 2021, 224, 121864. [Google Scholar] [CrossRef]
- Petersen, N.; Jensen, H.; Hansen, S.; Foss, S.; Snakenborg, D.; Pedersen-Bjergaard, S. On-chip electromembrane extraction. Microfluid. Nanofluidics 2010, 9, 881–888. [Google Scholar] [CrossRef]
- Zarghampour, F.; Yamini, Y.; Baharfar, M.; Javadian, G.; Faraji, M. On-chip electromembrane extraction followed by sensitive digital image-based colorimetry for determination of trace amounts of Cr (vi). Anal. Methods 2020, 12, 483–490. [Google Scholar] [CrossRef]
- Xu, Y.; Bai, H.; Lu, G.; Li, C.; Shi, G. Flexible graphene films via the filtration of water-soluble noncovalent functionalized graphene sheets. J. Am. Chem. Soc. 2008, 130, 5856–5857. [Google Scholar] [CrossRef]
- Ramezani, A.M.; Yamini, Y. Electrodeposition of poly-ethylenedioxythiophene-graphene oxide nanocomposite in a stainless steel tube for solid phase microextraction of letrozole in plasma samples. J. Sep. Sci. 2020, 43, 4338–4346. [Google Scholar] [CrossRef]
- Li, D.; Liu, M.; Zhan, Y.; Su, Q.; Zhang, Y.; Zhang, D. Electrodeposited poly(3,4-ethylenedioxythiophene) doped with graphene oxide for the simultaneous voltammetric determination of ascorbic acid, dopamine and uric acid. Microchim. Acta 2020, 187, 94. [Google Scholar] [CrossRef]
- Lehtimäki, S.; Suominen, M.; Damlin, P.; Tuukkanen, S.; Kvarnström, C.; Lupo, D. Preparation of Supercapacitors on Flexible Substrates with Electrodeposited PEDOT/Graphene Composites. ACS Appl. Mater. Interfaces 2015, 7, 22137–22147. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, S.H.; Jiang, H.Y.; Zeng, H.; Wu, R.M.; Chen, H.; Gao, Y.F.; Huang, Y.Y.; Bai, H.L. Properties of carbon black-PEDOT composite prepared via in-situ chemical oxidative polymerization. E-Polymers 2019, 19, 61–69. [Google Scholar] [CrossRef]
- Zhao, G.; Ren, X.; Gao, X.; Tan, X.; Li, J.; Chen, C.; Huang, Y.; Wang, X. Removal of Pb(II) ions from aqueous solutions on few-layered graphene oxide nanosheets. Dalton Trans. 2011, 40, 10945. [Google Scholar] [CrossRef]
- Bernal, V.; Erto, A.; Giraldo, L.; Moreno-Piraján, J. Effect of solution pH on the adsorption of paracetamol on chemically modified activated carbons. Molecules 2017, 22, 1032. [Google Scholar] [CrossRef]
- Gjelstad, A.; Rasmussen, K.E.; Pedersen-Bjergaard, S. Simulation of flux during electro-membrane extraction based on the Nernst–Planck equation. J. Chromatogr. A 2007, 1174, 104–111. [Google Scholar] [CrossRef]
- Dos Santos, M.F.; Ferri, C.C.; Seulin, S.C.; Leyton, V.; Pasqualucci, C.A.G.; Muñoz, D.R.; Yonamine, M. Determination of antidepressants in whole blood using hollow-fiber liquid-phase microextraction and gas chromatography–mass spectrometry. Forensic Toxicol. 2014, 32, 214–224. [Google Scholar] [CrossRef]
- Wan Ibrahim, W.N.; Sanagi, M.M.; Hanapi1, N.S.M.; Hadzir1, N.M.; Yahaya, N.; Kamaruzaman, S. Agarose-chitosan-integrated multiwalled carbon nanotubes film solid phase microextraction combined with high performance liquid chromatography for the determination of tricyclic antidepressant drugs in aqueous samples. Malaysian J. Anal. Sci. 2020, 24, 33–41. Available online: http://pkukmweb.ukm.my/mjas (accessed on 19 November 2022).
- Ito, R.; Ushiro, M.; Takahashi, Y.; Saito, K.; Ookubo, T.; Iwasaki, Y.; Nakazawa, H. Improvement and validation the method using dispersive liquid–liquid microextraction with in situ derivatization followed by gas chromatography–mass spectrometry for determination of tricyclic antidepressants in human urine samples. J. Chromatogr. B 2011, 879, 3714–3720. [Google Scholar] [CrossRef]
- Shamsayei, M.; Yamini, Y.; Asiabi, H. Polythiophene/graphene oxide nanostructured electrodeposited coating for on-line electrochemically controlled in-tube solid-phase microextraction. J. Chromatogr. A 2016, 1475, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.N.; Gonçalves, O.C.; Ahmad, S.M.; Schneider, J.K.; Krause, L.C.; Neng, N.R.; Caramão, E.B.; Nogueira, J.M.F. Application of bar adsorptive microextraction for the determination of levels of tricyclic antidepressants in urine samples. Molecules 2021, 26, 3101. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, H.; Banihashemi, S.; Zandian, F.K. Microextraction of antidepressant drugs into syringes packed with a nanocomposite consisting of polydopamine, silver nanoparticles and polypyrrole. Microchim. Acta 2015, 183, 195–202. [Google Scholar] [CrossRef]
- Kamaruzaman, S.; Sanagi, M.M.; Yahaya, N.; Wan Ibrahim, W.A.; Endud, S.; Wan Ibrahim, W.N. Magnetic micro-solid-phase extraction based on magnetite-MCM-41 with gas chromatography-mass spectrometry for the determination of antidepressant drugs in biological fluids. J. Sep. Sci. 2017, 40, 4222–4233. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | LOD (µg L−1) | LDR (µg L−1) | R2 | Inter-Day RSD (%, n = 3) | Intra-Day RSD (%, n = 3) | Fiber-to-Fiber RSD (%, n = 3) |
|---|---|---|---|---|---|---|
| Amitrptyline | 0.010 | 0.025–500 | 0.9989 | 4.5 | 5.3 | 7.2 |
| Nortriptyline | 0.010 | 0.025–500 | 0.9987 | 3.2 | 4.9 | 6.8 |
| Imipramine | 0.005 | 0.010–500 | 0.9996 | 4.3 | 2.9 | 5.3 |
| Desipramine | 0.010 | 0.025–1000 | 0.9986 | 5.1 | 5.4 | 6.1 |
| Maprotiline | 0.025 | 1.000–250 | 0.9984 | 4.8 | 6.2 | 8.4 |
| Sertraline | 0.005 | 0.010–500 | 0.9991 | 5.4 | 5.7 | 7.6 |
| Analytical Method | Analytes | LOD (µg L−1) | LDR (µg L−1) | RSD (%) | Ref. |
|---|---|---|---|---|---|
| HF-LPME-GC–MS 1 | 8 TCAs (Ami, Nor, Imi, Des) | 10.0 | 20.0–120.0 | 0.6–9.7 | [39] |
| SPME-HPLC–UV 2 | Ami, Chl 3 | 3.13–4.19 | 10–500 | ≤6.85 | [40] |
| DLLME-GC–MS 4 | 5 TCAs (Ami, Nor, Imi, Des) | 0.5–2.0 | 2.0–100 | 2.0–9.9 | [41] |
| EME-GC–FID | Imi, Clo 5 | 0.35–0.70 | 2–1500 | 8.0–8.5 | [17] |
| EC-IT-SPME-HPLC-UV 6 | Ami, Nor, Imi, Dex 7 | 0.3–0.5 | 0.70–200 | ≤8 | [42] |
| BAμE-GC–MS 8 | 6 TCAs (Am, Imi) | 0.2–1.6 | 10.0–1000 | ≤13.9 | [43] |
| MEPS-GC–MS 9 | Ami, Imi | 0.03–0.05 | 0.1–500 | ≤9.0 | [44] |
| Mμ-SPE-GC–MS 10 | Ami, Chl | 0.008–0.010 | 0.05–500 | ≤7.9 | [45] |
| On-chip EM-SPME-GC–MS | Ami, Nor, Imi, Des, Map, Ser | 0.01–0.25 | 0.5–500 | 4.5–8.1 | This work |
| Analyte | Creal (µg L−1) | Cadd (µg L−1) | Cfound (µg L−1) | RR i (%) | RSD (%) |
|---|---|---|---|---|---|
| Amitriptyline | Nd ii | 10.0 | 9.3 | 92.7 | 2.1 |
| 30.0 | 31.0 | 103.3 | 3.7 | ||
| Nortriptyline | Nd | 10.0 | 9.7 | 97.0 | 2.9 |
| 30.0 | 27.4 | 91.3 | 3.6 | ||
| Imipramine | Nd | 10.0 | 9.5 | 94.8 | 4.5 |
| 30.0 | 30.6 | 102.1 | 5.3 | ||
| Desipramine | Nd | 10.0 | 9.6 | 96.4 | 2.7 |
| 30.0 | 29.2 | 97.3 | 1.9 | ||
| Maprotiline | Nd | 10.0 | 9.9 | 98.6 | 4.6 |
| 30.0 | 31.1 | 103.6 | 3.4 | ||
| Sertraline | 32.0 | 10.0 | 41.3 | 93.2 | 5.7 |
| 30.0 | 62.7 | 102.4 | 4.4 |
| Analyte | Creal (µg L−1) | Cadd (µg L−1) | Cfound (µg L−1) | RR (%) | RSD (%) |
|---|---|---|---|---|---|
| Amitriptyline | Nd | 25.0 | 24.7 | 98.8 | 2.3 |
| 50.0 | 50.6 | 101.2 | 3.9 | ||
| Nortriptyline | Nd | 25.0 | 24.4 | 97.6 | 5.3 |
| 50.0 | 48.5 | 97.0 | 4.4 | ||
| Imipramine | 47.2 | 25.0 | 72.7 | 102.0 | 2.3 |
| 50.0 | 97.1 | 99.8 | 1.9 | ||
| Desipramine | Nd | 25.0 | 24.1 | 96.4 | 4.6 |
| 50.0 | 48.9 | 97.8 | 5.6 | ||
| Maprotiline | Nd | 25.0 | 24.3 | 97.2 | 4.7 |
| 50.0 | 48.3 | 96.6 | 6.3 | ||
| Sertraline | Nd | 25.0 | 26.3 | 105.2 | 1.1 |
| 50.0 | 49.8 | 99.6 | 2.3 |
| Analyte | Creal (µg L−1) | Cadd (µg L−1) | Cfound (µg L−1) | RR (%) | RSD (%) |
|---|---|---|---|---|---|
| Amitriptyline | Nd | 1.0 | 1.0 | 103.1 | 2.1 |
| 50.0 | 49.6 | 99.2 | 3.6 | ||
| 250.0 | 242.5 | 97.0 | 2.7 | ||
| Nortriptyline | Nd | 1.0 | 1.0 | 96.9 | 4.2 |
| 50.0 | 49.1 | 98.2 | 4.5 | ||
| 250.0 | 242.8 | 97.1 | 3.9 | ||
| Imipramine | Nd | 1.0 | 1.0 | 98.5 | 1.3 |
| 50.0 | 52.1 | 104.2 | 2.2 | ||
| 250.0 | 249.5 | 99.8 | 2.4 | ||
| Desipramine | Nd | 1.0 | 1.0 | 96.3 | 4.2 |
| 50.0 | 47.7 | 95.4 | 3.6 | ||
| 250.0 | 241.8 | 96.7 | 4.5 | ||
| Maprotiline | Nd | 1.0 | 0.9 | 94.6 | 5.2 |
| 50.0 | 47.7 | 95.3 | 4.4 | ||
| 250.0 | 237.3 | 94.9 | 3.9 | ||
| Sertraline | Nd | 1.0 | 1.0 | 99.9 | 1.1 |
| 50.0 | 50.6 | 101.1 | 2.6 | ||
| 250.0 | 247.3 | 98.9 | 1.9 |
| Analyte | Creal (µg L−1) | Cadd (µg L−1) | Cfound (µg L−1) | RR (%) | RSD (%) |
|---|---|---|---|---|---|
| Amitriptyline | Nd | 1.0 | 1.0 | 96.1 | 8.1 |
| 50.0 | 47.1 | 94.2 | 3.6 | ||
| 250.0 | 258.5 | 103.4 | 6.7 | ||
| Nortriptyline | Nd | 1.0 | 0.9 | 93.0 | 4.3 |
| 50.0 | 50.9 | 101.7 | 5.6 | ||
| 250.0 | 244.0 | 97.6 | 1.9 | ||
| Imipramine | Nd | 1.0 | 1.0 | 95.1 | 2.4 |
| 50.0 | 48.6 | 97.2 | 3.5 | ||
| 250.0 | 247.5 | 99.0 | 1.3 | ||
| Desipramine | Nd | 1.0 | 1.0 | 102.2 | 1.7 |
| 50.0 | 49.3 | 98.5 | 2.6 | ||
| 250.0 | 248.3 | 99.3 | 2.2 | ||
| Maprotiline | Nd | 1.0 | 1.0 | 97.8 | 3.8 |
| 50.0 | 52.7 | 105.4 | 4.1 | ||
| 250.0 | 235.5 | 94.2 | 3.9 | ||
| Sertraline | Nd | 1.0 | 1.0 | 97.6 | 1.1 |
| 50.0 | 49.6 | 99.1 | 2.0 | ||
| 250.0 | 254.3 | 101.7 | 2.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamani, R.; Yamini, Y. On-Chip Electromembrane Surrounded Solid Phase Microextraction for Determination of Tricyclic Antidepressants from Biological Fluids Using Poly(3,4-ethylenedioxythiophene)—Graphene Oxide Nanocomposite as a Fiber Coating. Biosensors 2023, 13, 139. https://doi.org/10.3390/bios13010139
Zamani R, Yamini Y. On-Chip Electromembrane Surrounded Solid Phase Microextraction for Determination of Tricyclic Antidepressants from Biological Fluids Using Poly(3,4-ethylenedioxythiophene)—Graphene Oxide Nanocomposite as a Fiber Coating. Biosensors. 2023; 13(1):139. https://doi.org/10.3390/bios13010139
Chicago/Turabian StyleZamani, Razieh, and Yadollah Yamini. 2023. "On-Chip Electromembrane Surrounded Solid Phase Microextraction for Determination of Tricyclic Antidepressants from Biological Fluids Using Poly(3,4-ethylenedioxythiophene)—Graphene Oxide Nanocomposite as a Fiber Coating" Biosensors 13, no. 1: 139. https://doi.org/10.3390/bios13010139