1. Introduction
Melanoma is an aggressive skin cancer with a high propensity to metastasise to the brain [
1]. Metastasis majorly occurs via the circulation through the blood–brain barrier (BBB). Here, the brain endothelial cells form the first physical barrier that needs to be breached by blood-borne cancer cells [
2,
3,
4]. We have recently shown using sophisticated biosensor technology, that the paracellular barrier strength of brain endothelial cells, is rapidly weakened by invasive melanoma cells [
5,
6]. Melanoma cells were shown to translocate rapidly to the paracellular borders of the endothelial cells, and progress to separate the junctional borders and traverse between the neighbouring endothelial cells. Importantly, the loss of barrier integrity was rapid and evident within 60 min of melanoma cell addition to the apical face of the endothelial cells. Such a fast effect suggested that melanoma cells are able to directly alter the molecular integrity of the paracellular junctional cleft. Theoretically, this can be mediated by a range of enzymes that catalyse the breakdown of proteins for various functions, such as cell and tissue remodelling, cell and tissue regulation and cell signalling, detailed below. These enzymes are proteases, which are classified into seven large families, based on their catalytic site residues, as aspartic, cysteine, serine, metallo, threonine, glutamic and asparagine peptidases [
7,
8].
One large metallopeptidase subfamily, called matrix metalloproteinases (MMPs), have been extensively studied for their importance in the melanoma metastatic cascade [
9,
10]. MMPs are also implicated in destabilising the neurovascular unit during intracerebral traumas [
11]. MMPs are zinc-dependant endopeptidases [
10,
12,
13] consisting at least of a pro-domain, catalytic domain and a highly conserved active site [
14]. Over twenty-five different MMPs have been identified and these are categorised into different functional classes [
9,
14,
15]. The classes define their predominant role as gelatinases, collagenases, stromelysins, matrilysins, metalloelastases, membrane-type proteases and others [
16,
17,
18]. Cumulatively, these molecules cleave various extracellular matrix (ECM) materials, and several may interact with integrins and adhesion molecules for optimal positioning and protease activity [
9]. These characteristics are essential in cancer, where ECM remodelling and regulation is a key mechanism for cancer growth, progression, and metastasis. A major cancer related MMP is the gelatinase MMP-2, which is often upregulated in melanoma [
19] and is closely associated with invasion and metastasis. One of the activators of MMP-2 is another membrane-type MT1-MMP [
20,
21] and once activated at the membrane, MMP-2 can remain membrane bound, be released into the surrounding environment or attach to integrins [
22] to degrade the ECM. The upregulation of MMP-2, with its membrane receptor at the leading front of the cell invadopodium, allows modulation of melanoma adhesion and spreading in an ECM environment [
23]. In addition, MMP-2 may also bind to αVβ3 integrin to facilitate migration at the primary site [
24]. MMP-2 expression is also significantly increased in the tumour tissue of patients with melanoma at the primary and secondary sites [
25]. MMP-2 mRNA and protein are expressed in human melanoma mouse xenografts and this expression positively correlates with melanoma aggressiveness [
26].
Other MMPs of interest include the collagenase MMP-1, stromelysin MMP-3 and gelatinase B MMP-9. MMP-1 activation is correlated with promoting melanoma progression into the more aggressive ventricle growth phase [
27,
28]. MMP-1 is upregulated in melanoma in vivo [
26] and both MMP-1 and -3 are associated with shorter disease-free survival [
29,
30]. MMP-3 has also been shown to increase the BBB permeability and extravasation of dyes in mouse models [
31]. Another example is MMP-9, which is suggested as a marker for treatment assessment in melanoma patients [
32] and with uveal melanoma [
33]. In mice, the addition of recombinant inhibitors of MMPs reduces the number of melanoma lung metastasis (but not the size of the metastasis), suggesting their importance in extravasation [
34]. Collectively, this makes MMPs a crucial family to investigate in melanoma disruption of the brain endothelium.
In addition to matrix metalloproteinases, a large repertoire of enzymatic families exists as ECM degrading molecules. A serine-based protease called seprase has been closely associated with melanoma migration through the endothelial monolayer in a Transwell system [
35]. Another serine protease, called kallikrein-related peptidase 6 (KLK6), was shown to increase melanoma invasion though a Matrigel scaffold when released by supporting stromal cells [
36]. Additionally, cathepsins, which are a family of lysosomal proteases, are highly implicated in cancer progression and growth, and lead to poor prognosis specifically in malignant melanoma [
37,
38,
39,
40]. Cathepsins cover many different types of proteases, but the cysteine based Cathepsin B, and L and aspartic based Cathepsin D, are most associated with an increased in vitro proliferation and progression of melanoma [
41,
42,
43]. One proposed mechanism is that Cathepsin B expression in melanoma stimulates fibroblast activation through a transforming growth factor β (TGFβ)-dependent pathway, and this stromal involvement increases melanoma traversal through the basement membrane; however, this acts only as an additive mechanism, as Cathepsin B also supports melanoma invasiveness without the presence of fibroblasts [
44]. Due to their multiple and dynamic roles, it was hypothesized that other existing proteases and protease families may also be directly or indirectly correlated with cancer invasion.
This study aimed to assess the involvement of a range of proteolytic enzymes in melanoma mediated brain endothelial barrier disruption. The objective was to identify targetable proteins in the blood, prior to melanoma extravasation past the brain endothelium. The prime suspects were MMPs, which were hypothesized to facilitate the melanoma mediated loss of brain endothelial barrier strength. The theory herein, was that blocking a variety of these molecules with inhibitors of a low nanomolar potency would decrease the ability of melanoma cells to invade at the endothelial cell junctions substantially, if the proteases were majorly involved in this process.
4. Discussion
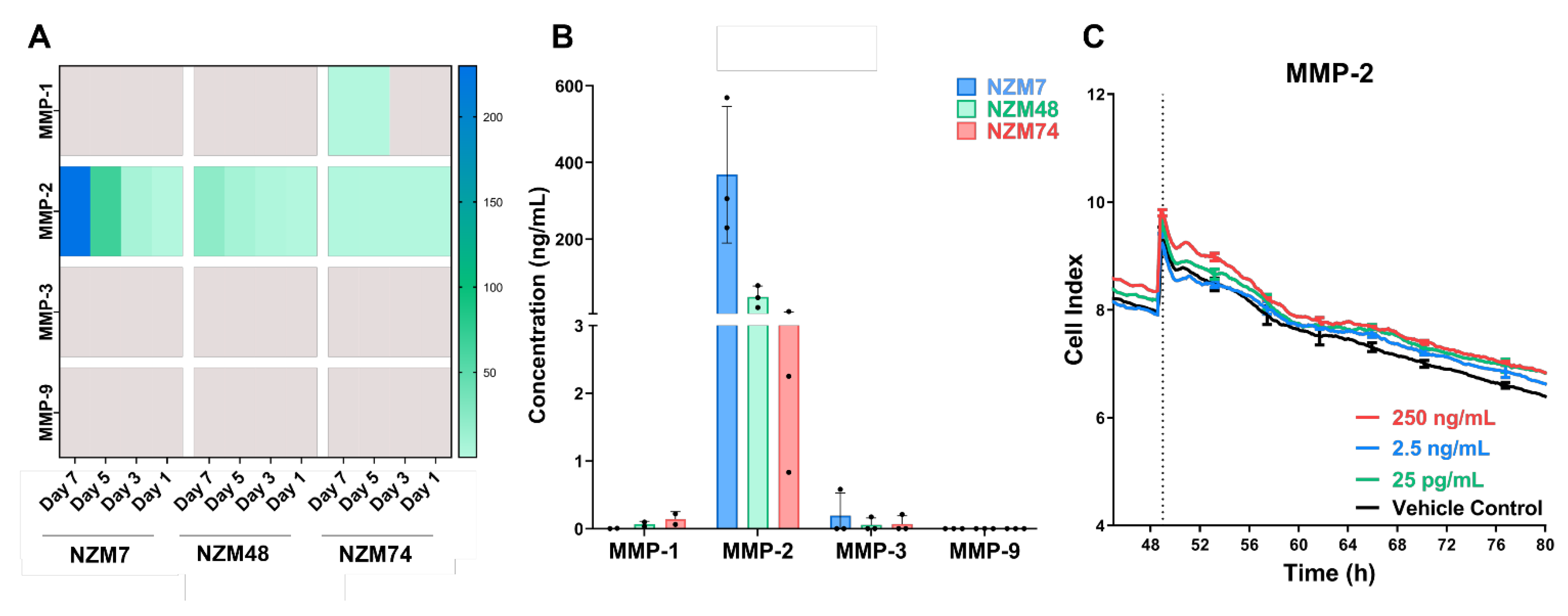
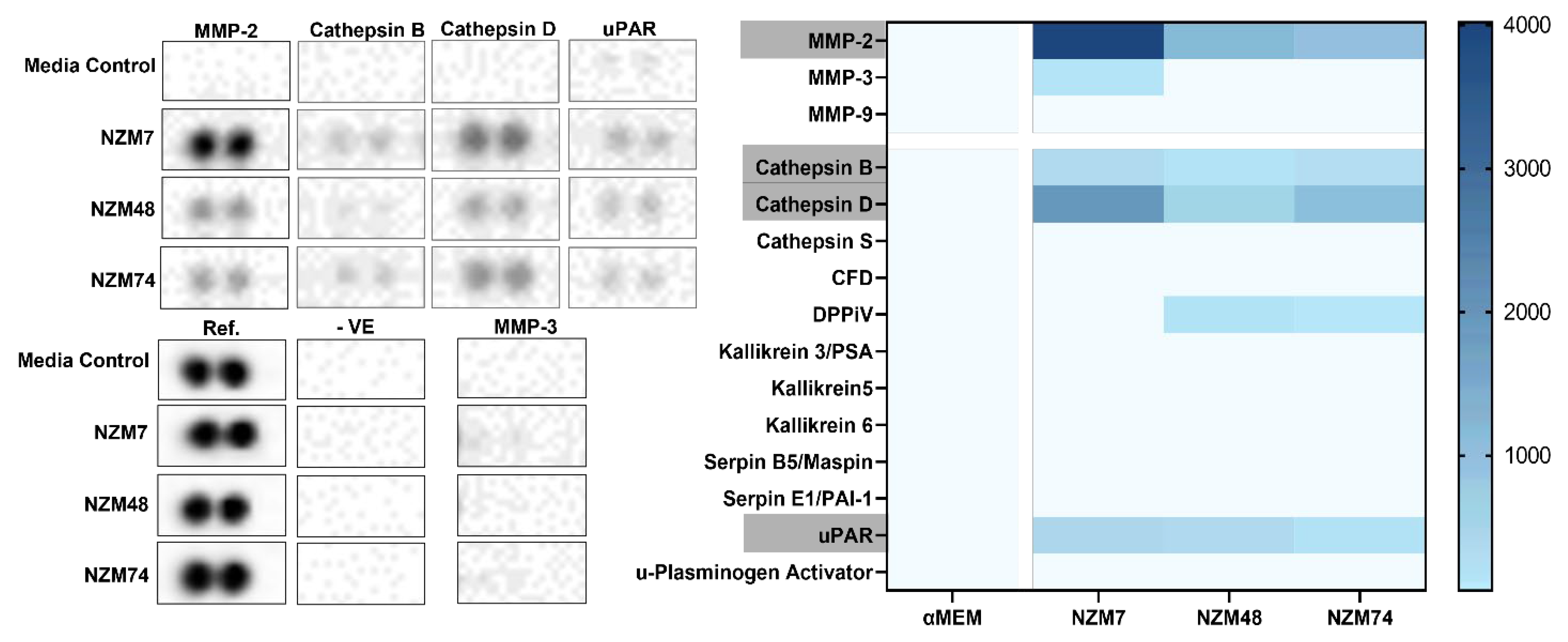
Herein, the involvement of proteases in melanoma mediated disruption of the brain endothelial cell barrier was assessed. The hypothesis was that various proteolytic enzymes, such as MMPs and serine-, cysteine- and aspartic- proteases, expressed by the melanoma lines facilitated the breakdown of junctional molecules to initiate disruption of the endothelial barrier. The assessed melanoma lines showed an expression of various proteases including MMP-2, Cathepsin B and Cathepsin D. MMPs -3 and -9, which are commonly correlated with melanoma metastases, were not detected in melanoma conditioned media, though these have conflicting roles in melanoma progression, with data that suggest both pro and anti-tumour activity [
30,
59,
60,
61]. Surprisingly, the urokinase plasminogen activator (uPA) was not detected in the conditioned media, although previous studies have shown that uPA mRNA is upregulated in metastatic cancer over benign nevi [
44]; however, the receptor uPAR was detected. uPAR is a co-factor for plasminogen activation and interacts with uPA to cleave plasminogen into its active form, plasmin. Plasmin is a serine protease that cleaves fibrin, functioning as a de-clotting agent. Interestingly, uPAR is typically a cell-surface molecule [
62] but can be cleaved in monocytes to act as a biological activator of chemotaxis and cell-adhesion [
63], which may explain its expression in melanoma conditioned media.
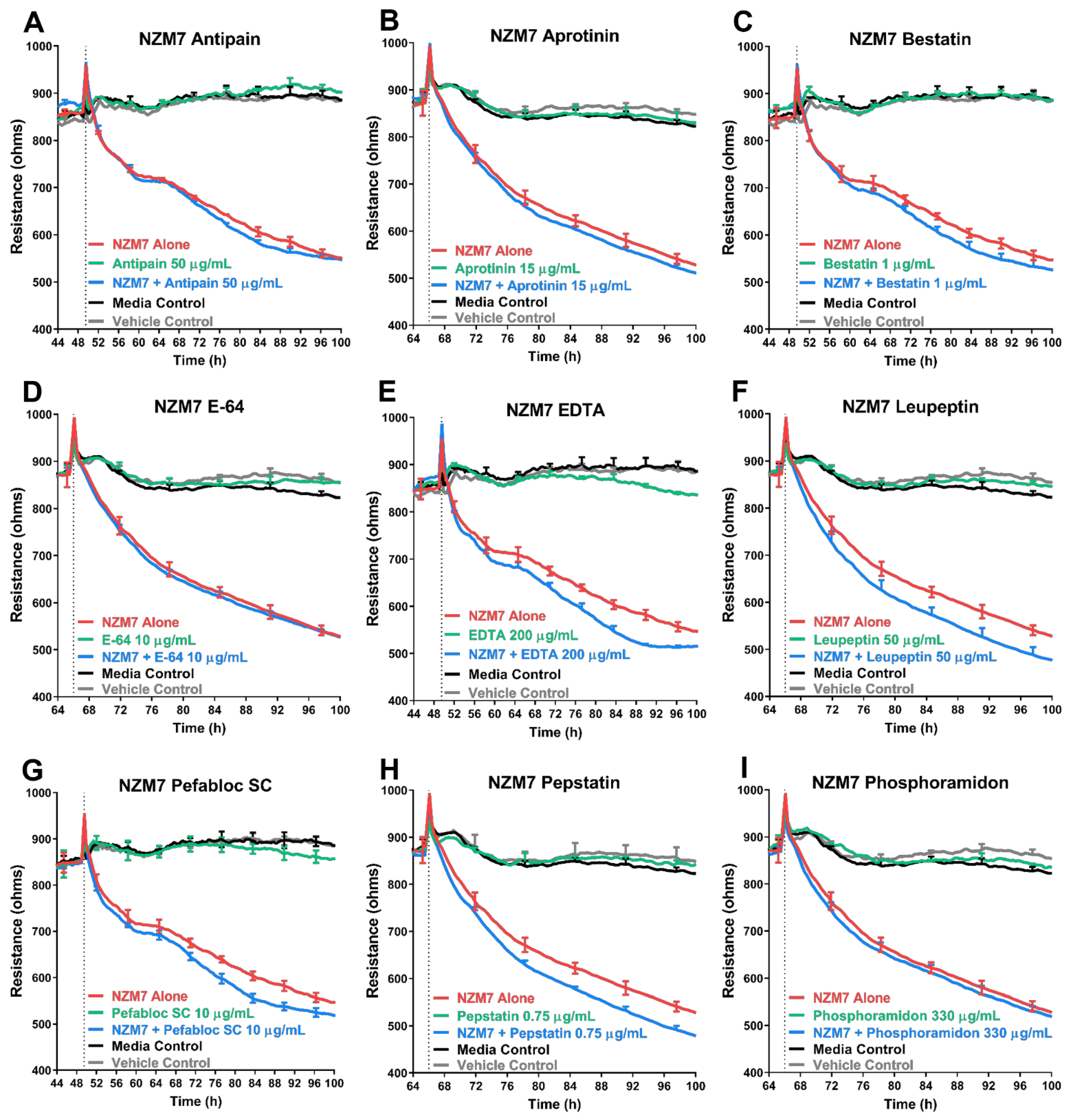
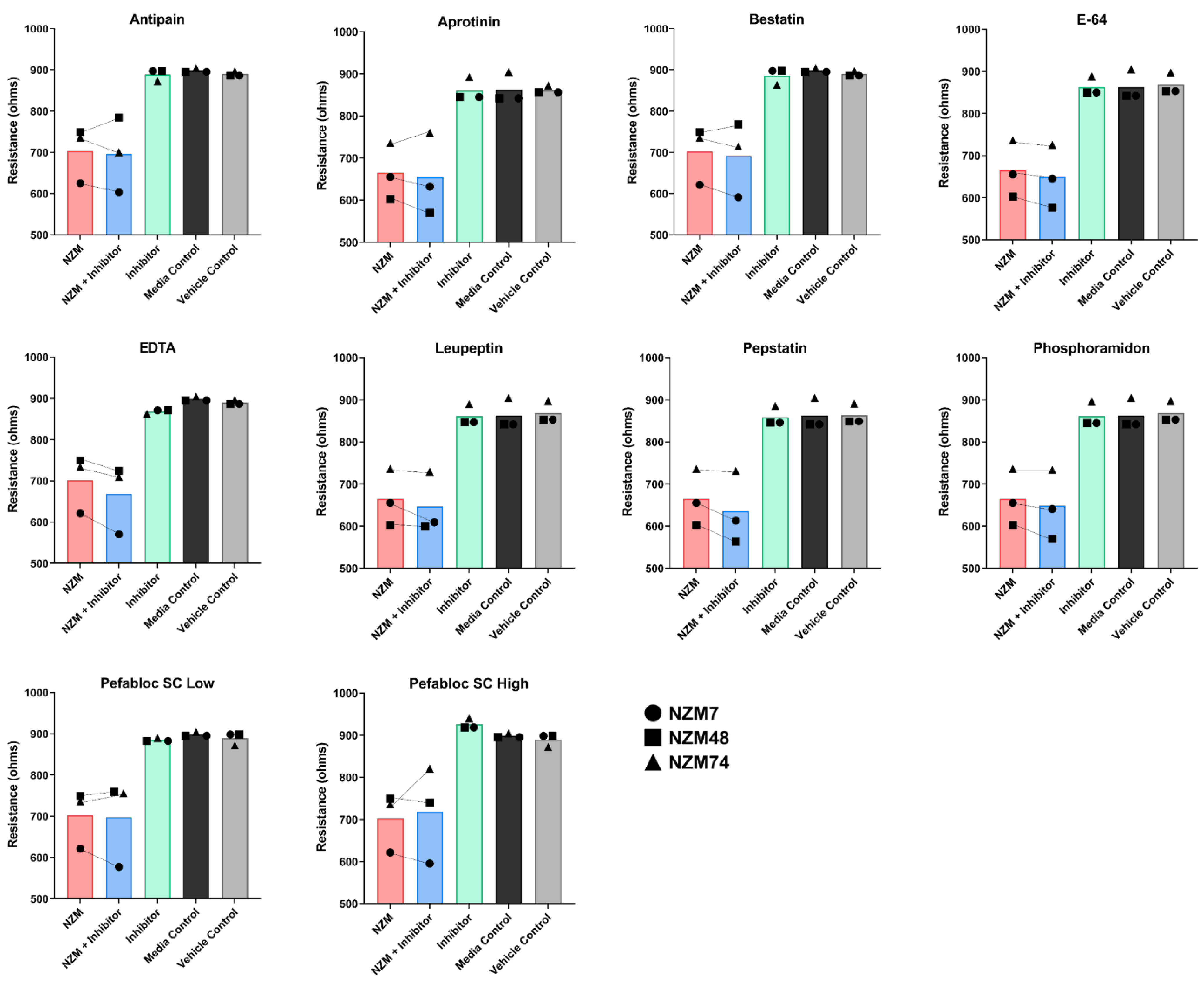
The addition of MMP-2 to the apical face of the brain endothelial monolayer showed no effect on the overall endothelial barrier function. The addition of broad-spectrum inhibitors to the melanoma cells also did not hinder the ability of the melanoma lines to disrupt the endothelial barrier and similar results were seen with a range of inhibitors of other proteases. An important point to note, was that the protease inhibitors were co-incubated with the melanoma cells and added to the endothelial cells as a melanoma-inhibitor cocktail. This was performed to ensure that the inhibitors were not only present on the melanoma alone but also at the invasive front on the apical surface of the endothelial cells, upon and after melanoma addition. The inhibitors were, therefore, expected to block some protease activity if it were majorly involved in mediating endothelial barrier disruption. The results indicate that this was not the case for any of the melanoma lines assessed.
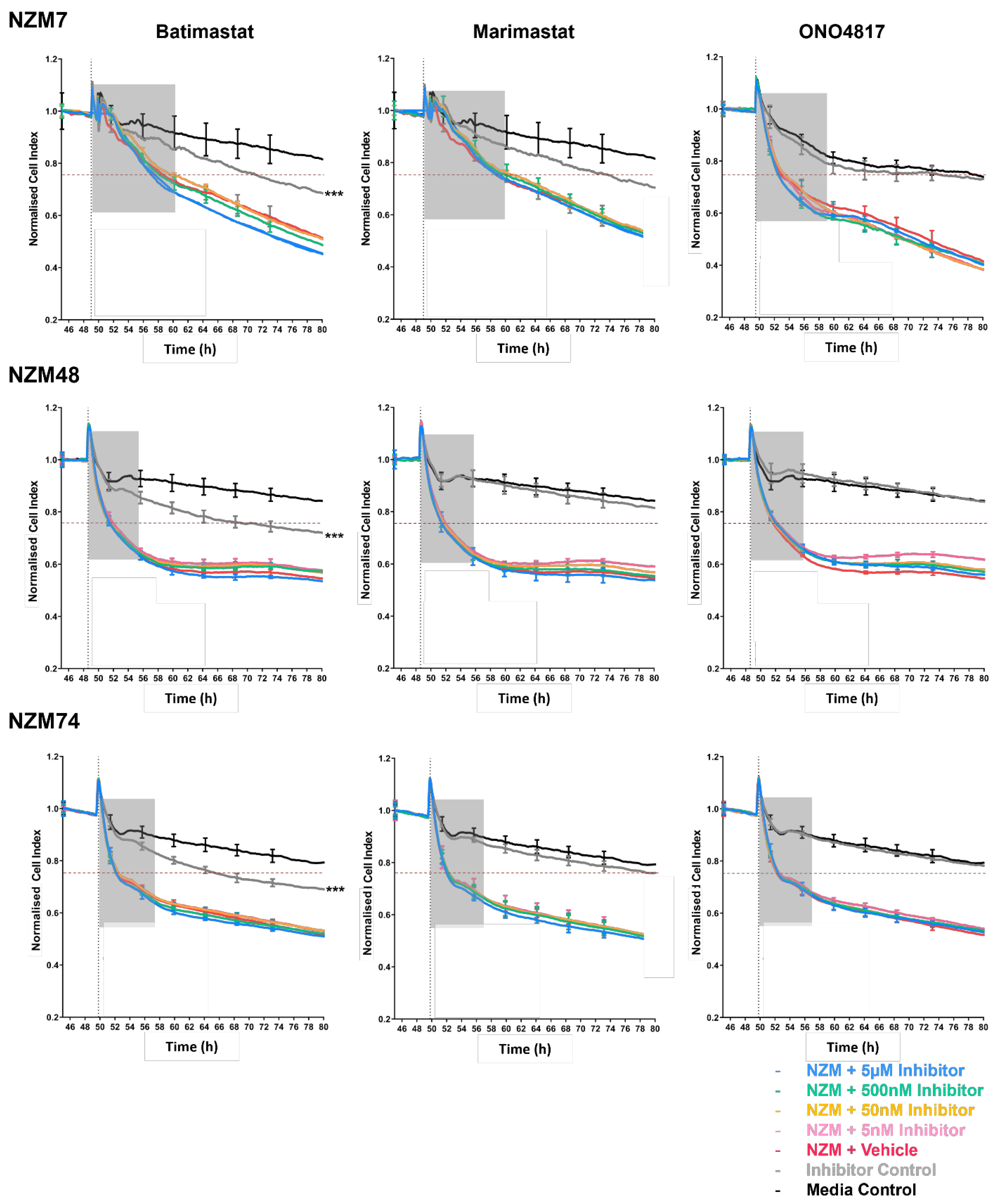
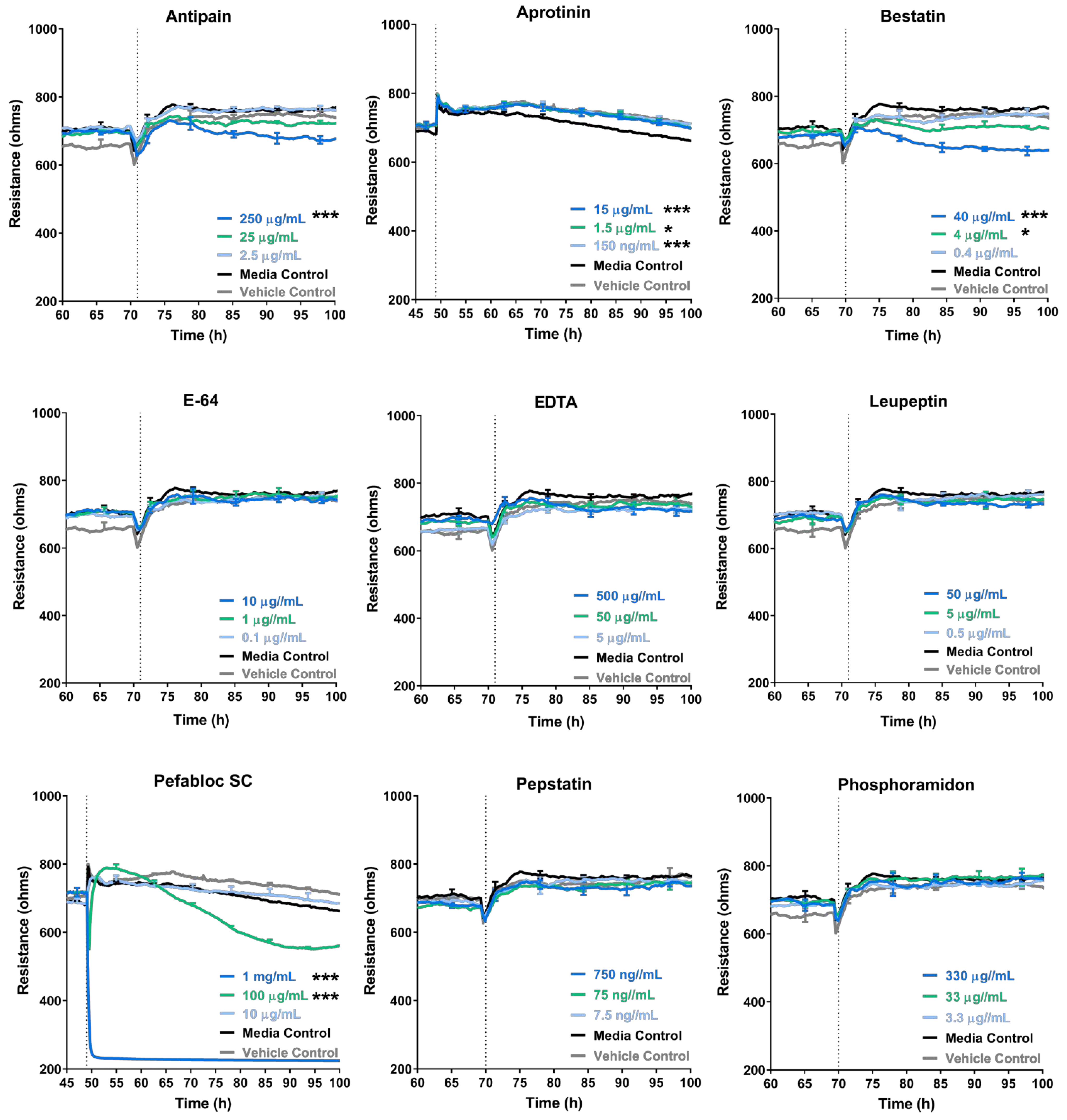
Intriguingly, the addition of some inhibitors such as batimastat, marimastat and Pefabloc SC, disrupted the endothelial barrier independent of the melanoma cells, which highlights that the inhibitors have an effect (which is potentially detrimental) on the endothelial barrier alone. Interpreting the specificities of the MMP inhibitors showed that there was no overlap of MMP inhibition specificity, exclusively between marimastat and batimastat; however, there were four MMPs (MMP-1, -2, -7 and -9), which were inhibited by all three inhibitors (
Supplemental Figure S5A). Interestingly, batimastat was found to be an extremely potent inhibitor of MMP-1 (IC
50: 3 nM, as described by the manufacturer, R&D Systems). Marimastat was also potent but less than the batimastat, whereas ONO4817, with an IC
50 value of 1600 nM, was the least potent. This trend is depicted in
Table 3, and also matched the barrier disruption trends in
Figure 3 and
Supplemental Figure S4, where the inhibitor batimastat disrupted the barrier the most. This, however, is not conclusive nor been tested in this paper, and it is likely that the high concentrations of drugs, in general, have a detrimental effect on the brain endothelial barrier. The effect, however, was not as large as what we see with the melanoma lines [
5], or with inflammatory cytokines [
48].
Cumulatively, these results suggested that although several proteases may affect the endothelial cells independently over time, the MMPs and other proteases do not make a reliable target for blocking melanoma metastasis at the locus of extravasation, which is at the apical face of the endothelial cells. It also suggests that MMPs likely do not influence the melanoma mediated endothelial barrier disruption we have previously seen in [
5]. These results were initially unexpected, as proteases have proven to facilitate melanoma metastases through the ECM at the primary tumour site to facilitate travel to the nearest blood vessel for intravasation into the circulation [
9,
64]. Melanoma protease expression is also correlated with a better chance of extravasation at the secondary site [
65] and there is corresponding evidence of melanoma migration through the endothelial monolayer being inhibited by protease inhibitors [
35]. In-depth investigation of the literature on protease mediated migration suggests that although proteases play an important role in melanoma migration through the ECM at the primary site, at the secondary site, proteases may only facilitate migration after the extravasation step which occurs past the endothelial monolayer and in the basement membrane and parenchyma [
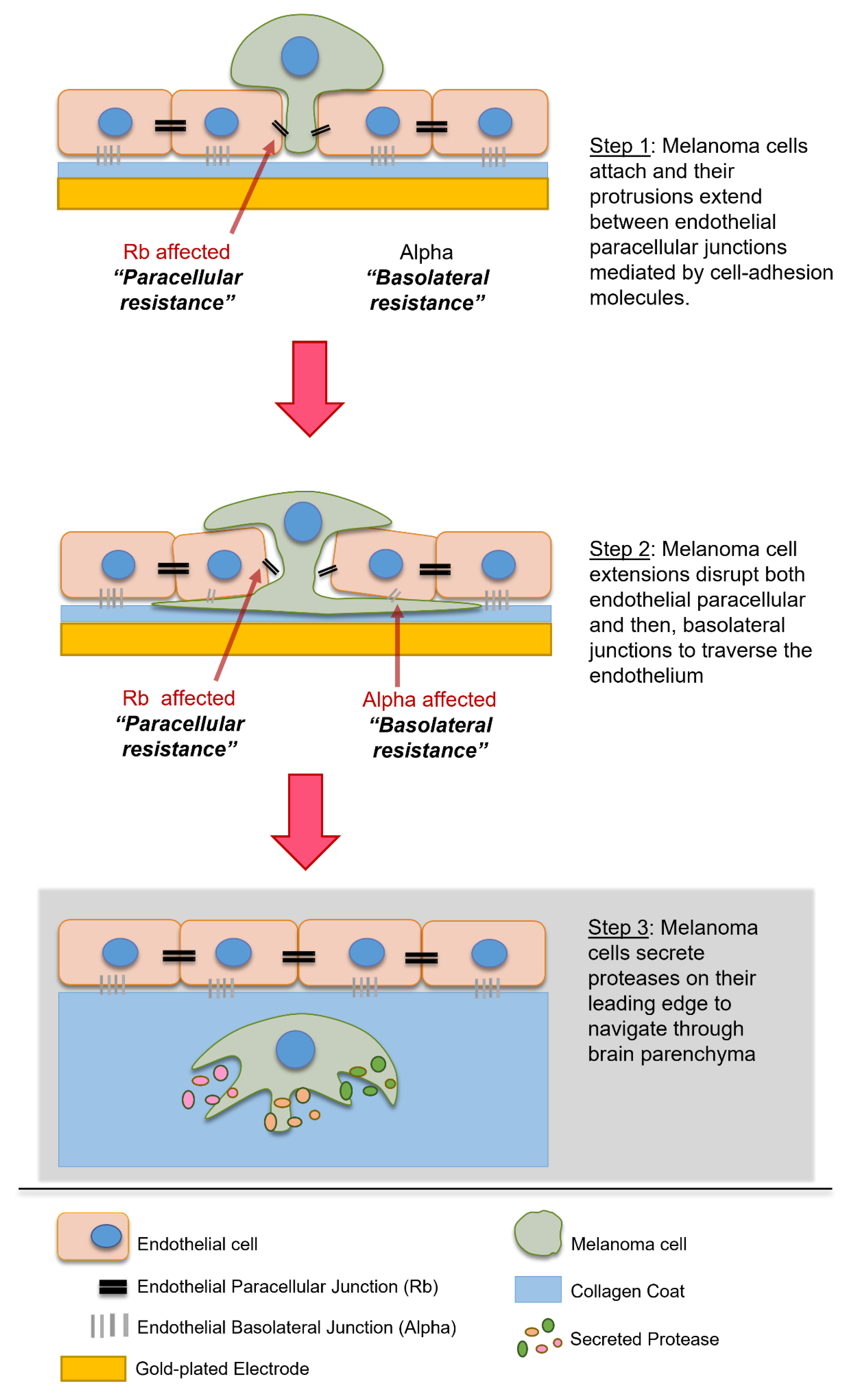
66], depicted in
Figure 8. This was deduced as most migration studies use Matrigel based scaffolds to assess invasion rather than cellular barriers, such as the vascular endothelium. In vivo studies suggest that proteases may also be important in regulating clotting agents and platelet interactions which are major support factors for circulating melanoma cells; therefore, it is likely that the correlation between a high expression of proteases and metastatic potential is attributed to (i) traversal through ECM at the primary site, (ii) survival in the circulation and (iii) traversal through the basement membrane after the more profound layers of the blood–brain barrier have been breached, but not attributed to extravasation at the site of the brain endothelial junctions which need to be disrupted first.
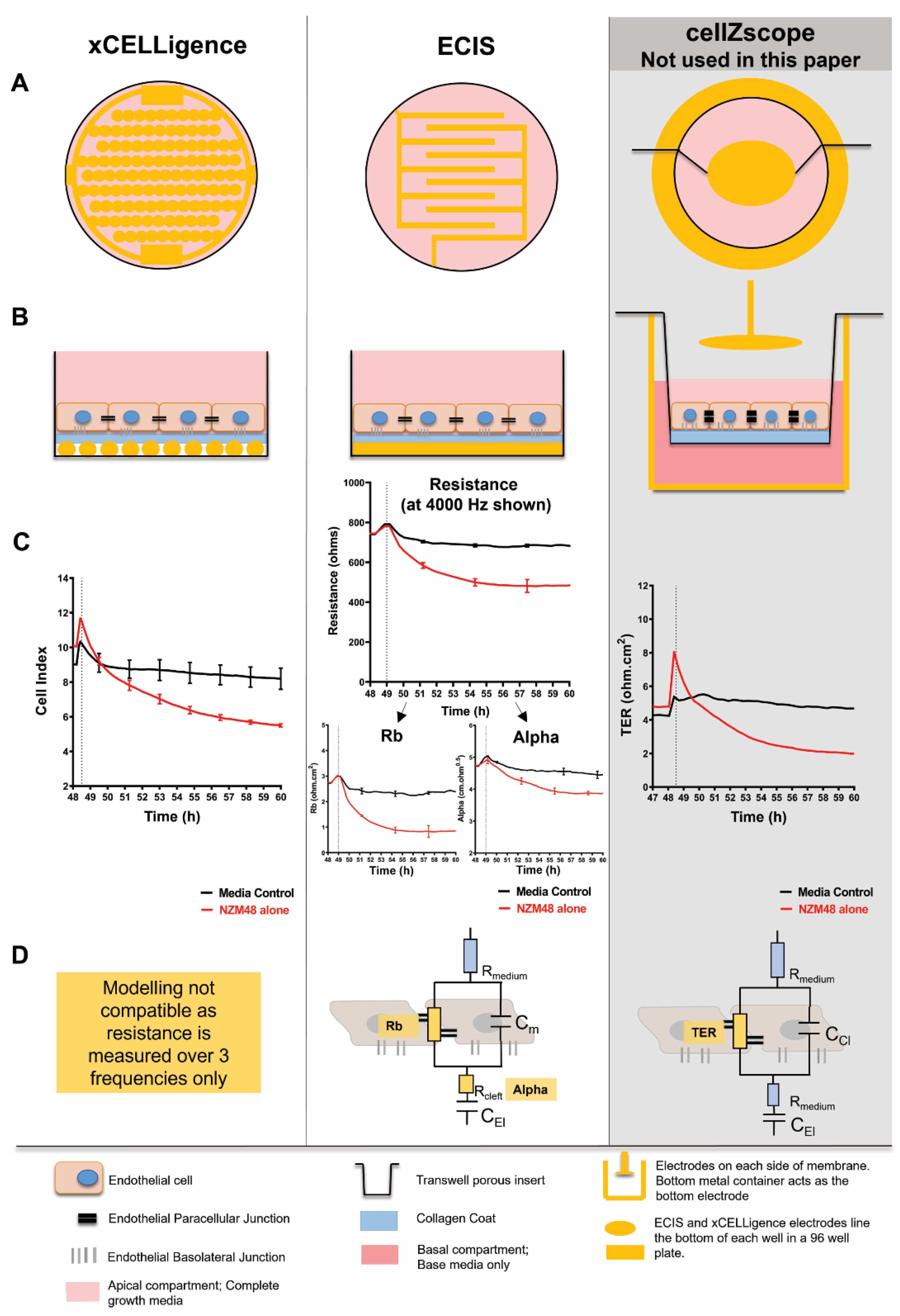
Additionally, it is important to establish reliable and relevant models when designing an experimental question. It can be proposed that there are differences in the cell models grown on flat surfaces and across Transwell systems as used by Fazakas, Wilhelm [
35] and this leads to disparities between cross-modal analyses. For this paper, high-throughput, real-time impedance biosensing was used to temporally assess the fast-acting mechanism occurring at the invasive front of melanoma extravasation. This front is critically on the apical face of the highly polarised brain endothelial cells, interfacing the blood. The sensitivity of impedance sensing allowed for the detection of very small changes occurring during the melanoma–endothelial interaction, and the addition of all our treatments to only the apical face of the endothelial cells allowed for spatial assessment of the effect of the proteases on the endothelial monolayer.
This was important as it revealed that the inhibition of proteases could not hinder melanoma mediated disruption of the endothelial barrier junctions. Although MMPs and other proteases may play an important role in aiding melanoma invasiveness past the endothelium, melanoma cells must still disrupt the endothelial barrier first to traverse through the paracellular space; therefore, future studies will assess the expression and effect of pre-existing key molecular players, present at the invasive front of the melanoma cells. This is particularly important due to the fast nature of the melanoma effect. If a link is found, studies must translate this research into sheer-based systems of a complete BBB, to better replicate the apical front of the brain endothelial barrier.
Brain-metastatic cancers have poor clinical outcomes [
67,
68,
69,
70] and are substantially involved in cancer treatment failure [
71,
72]. This provides the biological rationale to target metastasis in the initial process of extravasation, at the brain endothelium. Thereby, we need to identify and potentially block the metastatic mechanisms used by cancer cells, whilst they are still an accessible target in the blood. Consequently, there is an urgent need to understand the mechanisms of extravasation that can have a therapeutic role in brain metastases. In this paper, we propose that proteinases such as MMPs are not primarily involved in the melanoma mediated barrier disruption of brain endothelial cells and, therefore, do not make reliable targets for therapeutic intervention at the brain endothelial front.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






