Optimisation of an Electrochemical DNA Sensor for Measuring KRAS G12D and G13D Point Mutations in Different Tumour Types †



Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Sensor Development and Set-Up
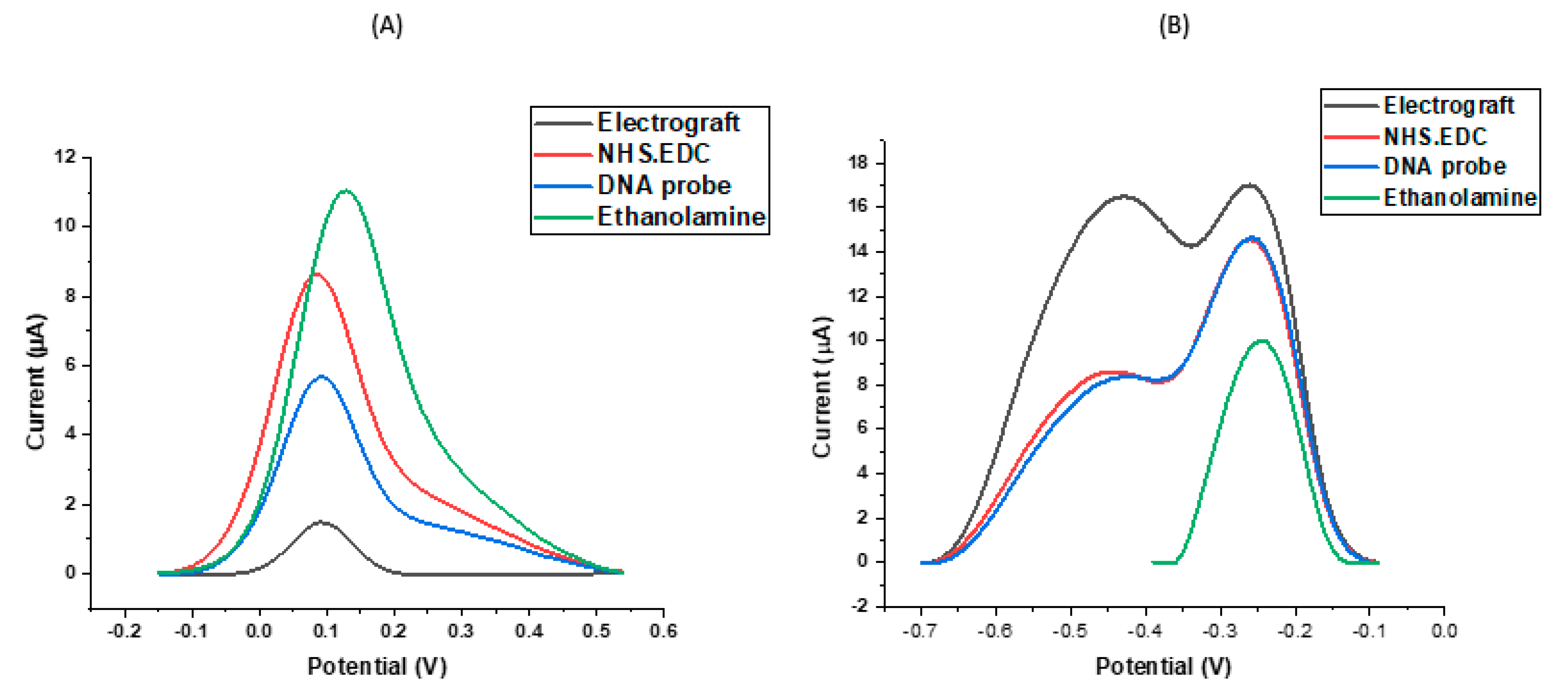
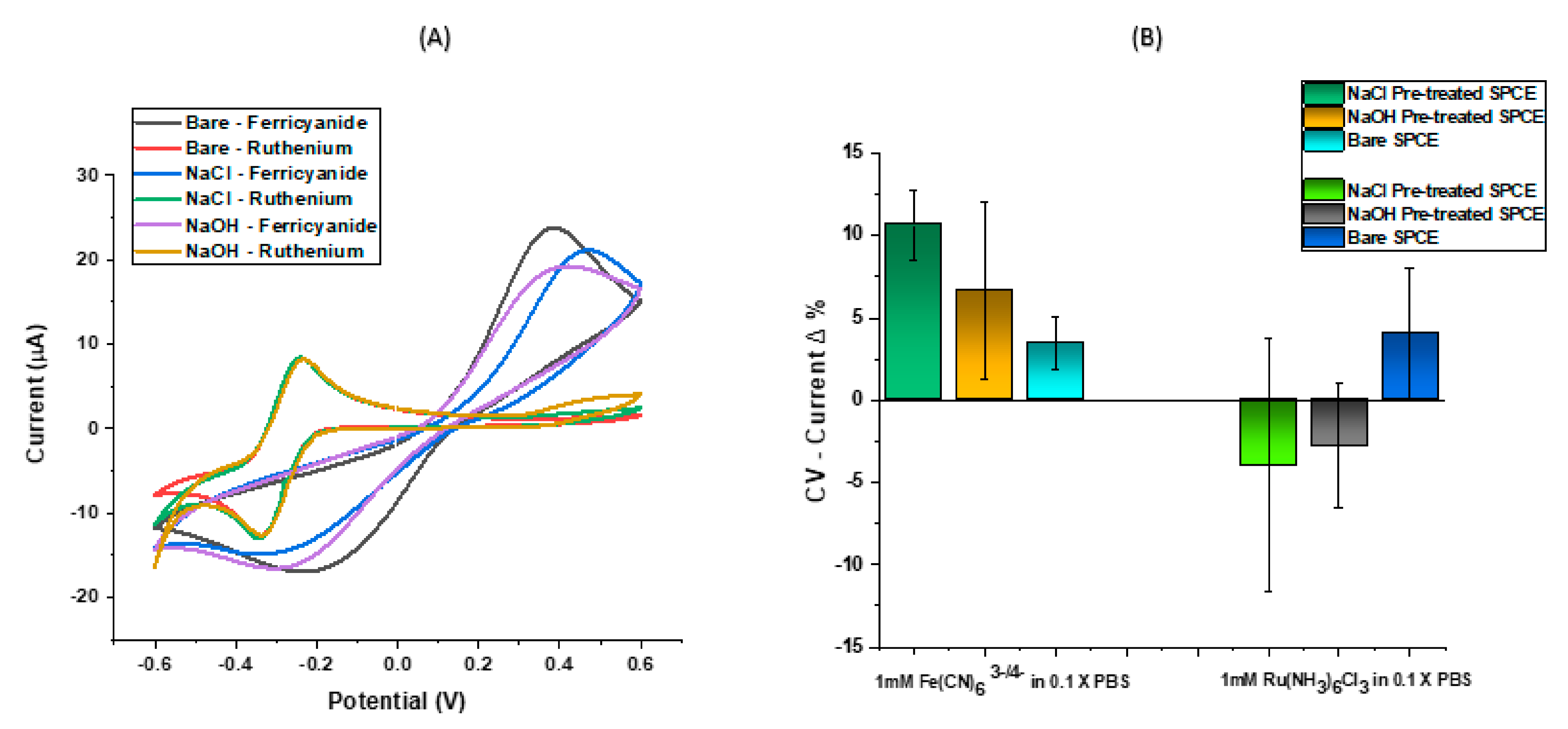
2.3. Electrode Preparation and Surface Functionalisation
2.4. Genomic DNA Sample Preparation, DNA Probe Design, and Sample Amplification
3. Results and Discussion
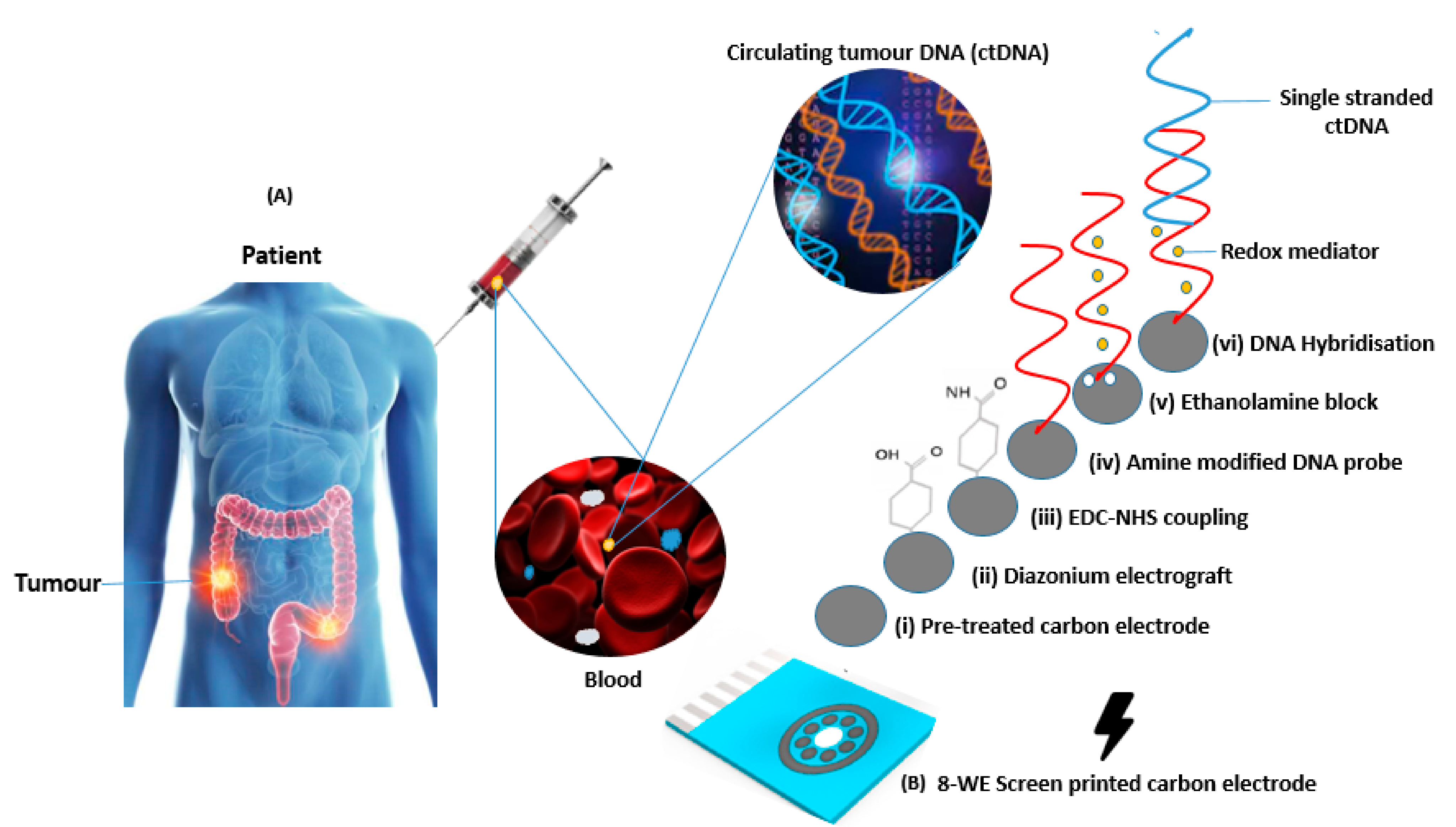
3.1. Assay Workflow and Development
3.2. DNA Sensor Hybridisation Specificity
3.3. KRAS G13D Amplification and Negative Control
3.4. Concentration Dose Response
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Attoye, B.; Baker, M.; Pou, C.; Thomson, F.; Corrigan, D.K. Electrochemical DNA Detection Methods to Measure Circulating Tumour DNA for Enhanced Diagnosis and Monitoring of Cancer. Proceedings 2020, 60, 15. [Google Scholar]
- National Cancer Institute. Genetic Changes and Cancer. 2019. Available online: https://www.cancer.gov/about-cancer/causes-prevention/genetics (accessed on 12 March 2020).
- Othman, E.; Wang, J.; Sprague, B.L.; Rounds, T.; Ji, Y.L.; Herschorn, S.D.; Wood, M.E. Comparison of false positive rates for screening breast magnetic resonance imaging (MRI) in high risk women performed on stacked versus alternating schedules. Springerplus 2015, 4, 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyamala, K.; Girish, H.C.; Murgod, S. Risk of tumor cell seeding through biopsy and aspiration cytology. J. Int. Soc. Prev. Community Dent. 2014, 4, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vendrell, J.A.; Taviaux, S.; Béganton, B.; Godreuil, S.; Audran, P.; Grand, D.; Clermont, E.; Serre, I.; Szablewski, V.; Coopman, P.; et al. Detection of known and novel ALK fusion transcripts in lung cancer patients using next-generation sequencing approaches. Sci. Rep. 2017, 7, 12510. [Google Scholar] [CrossRef] [Green Version]
- Gorgannezhad, L.; Umer, M.; Islam, N.; Nguyen, N.; Shiddiky, M.J.A. Lab on a Chip Circulating tumor DNA and liquid biopsy: Opportunities, challenges, and recent advances in detection technologies. Lab Chip 2018, 1174–1196. [Google Scholar] [CrossRef]
- Chu, Y.; Cai, B.; Ma, Y.; Zhao, M.; Ye, Z.; Huang, J. Highly sensitive electrochemical detection of circulating tumor DNA based on thin-layer MoS2/graphene composites. RSC Adv. 2016, 6, 22673–22678. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Taly, V.; Pekin, D.; Benhaim, L.; Kotsopoulos, S.K.; Corre, D.L.; Li, X.; Atochin, I.; Link, D.R.; Griffiths, A.D.; Pallier, K.; et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin. Chem. 2013, 59, 1722–1731. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223. [Google Scholar] [CrossRef]
- AACR Project Genie Consortium. AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Kulemann, B.; Rösch, S.; Seifert, S.; Timme, S.; Bronsert, P.; Seifert, G.; Martini, V.; Kuvendjiska, J.; Glatz, T.; Hussung, S.; et al. Pancreatic cancer: Circulating Tumor Cells and Primary Tumors show Heterogeneous KRAS Mutations. Sci. Rep. 2017, 7, 4510. [Google Scholar] [CrossRef] [Green Version]
- Shackelford, R.E.; Whitling, N.A.; McNab, P.; Japa, S.; Coppola, D. KRAS Testing: A Tool for the Implementation of Personalized Medicine. Genes Cancer 2012, 3, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Strausberg, R.L.; Buetow, K.H.; Emmert-Buck, M.R.; Klausner, R.D. The Cancer Genome Anatomy Project: Building an annotated gene index. Trends Genet. 2000, 16, 103–106. [Google Scholar] [CrossRef]
- Imyanitov, E.N. Gene polymorphisms, apoptotic capacity and cancer risk. Hum. Genet. 2009, 125, 239–246. [Google Scholar] [CrossRef]
- Kelley, S.O. What Are Clinically Relevant Levels of Cellular and Biomolecular Analytes? ACS Sens. 2017, 2, 193–197. [Google Scholar] [CrossRef]
- Crossley, L.; Attoye, B.; Vezza, V.; Blair, E.; Corrigan, D.K.; Hannah, S. Establishing a Field-Effect Transistor Sensor for the Detection of Mutations in the Tumour Protein 53 Gene (TP53)—An Electrochemical Optimisation Approach. Biosensors 2019, 9, 141. [Google Scholar] [CrossRef] [Green Version]
- Russell, C.; Ward, A.C.; Vezza, V.; Hoskisson, P.; Alcorn, D. Biosensors and Bioelectronics Development of a needle shaped microelectrode for electrochemical detection of the sepsis biomarker interleukin-6 (IL-6) in real time. Biosens. Bioelectron. 2019, 126, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, A.; Ward, A.C. Analytical Methods Electrochemical detection of oxacillin resistance with SimpleStat: A low cost integrated potentiostat and sensor platform. Anal. Methods 2019, 11, 1958–1965. [Google Scholar] [CrossRef] [Green Version]
- Dorothee, G.; Robert, M.; Janos Voros, E.R. Electrochemical Biosensors—Sensor Principles and Architectures. Sensors 2008, 8, 1400–1458. [Google Scholar]
- Shoaie, N.; Forouzandeh, M.; Omidfar, K. Voltammetric determination of the Escherichia coli DNA using a screen-printed carbon electrode modified with polyaniline and gold nanoparticles. Microchim. Acta 2018, 185, 1–9. [Google Scholar] [CrossRef]
- Li, P.Q.; Piper, A.; Schmueser, I.; Mount, A.R.; Corrigan, D.K. Correction: Impedimetric measurement of DNA–DNA hybridisation using microelectrodes with different radii for detection of methicillin resistant Staphylococcus aureus (MRSA). Analyst 2017, 142, 1946–1952. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Arrigan, D.W.M.; Silvester, D.S. Sensing and Bio-Sensing Research Mechanical polishing as an improved surface treatment for platinum screen-printed electrodes. Sens. BioSens. Res. 2016, 9, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Waldvogel, S.R.; Jiang, X. Electrochemistry of Carbon Dioxide on Carbon Electrodes. ACS Appl. Mater. Interfaces 2016. [Google Scholar] [CrossRef]
- Iniesta, J.; Thakur, B.; Carmo, R.; Banks, C.E. Can the mechanical activation (polishing) of screen-printed electrodes enhance their electroanalytical response? Analyst 2016, 141, 2791–2799. [Google Scholar] [CrossRef] [Green Version]
- Mahmoodi, P.; Rezayi, M.; Rasouli, E.; Avan, A.; Gholami, M. Early-stage cervical cancer diagnosis based on an ultra-sensitive electrochemical DNA nanobiosensor for HPV-18 detection in real samples. J. Nanobiotechnol. 2020, 1–12. [Google Scholar] [CrossRef] [Green Version]
- González-sánchez, M.I.; Gómez-monedero, B.; Agrisuelas, J.; Iniesta, J.; Valero, E. Electrochemistry Communications Highly activated screen-printed carbon electrodes by electrochemical treatment with hydrogen peroxide. Electrochem. Commun. 2018, 91, 36–40. [Google Scholar] [CrossRef]
- Wei, H.; Sun, J.J.; Xie, Y.; Lin, C.G.; Wang, Y.M.; Yin, W.H.; Chen, G.N. Enhanced electrochemical performance at screen-printed carbon electrodes by a new pretreating procedure. Anal. Chim. Acta 2007, 588, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Du, C.X.; Han, L.; Dong, S.L.; Li, L.H.; Wei, Y. A novel procedure for fabricating flexible screen-printed electrodes with improved electrochemical performance. IOP Conf. Ser. Mater. Sci. Eng. 2016, 137, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Elgrishi, N.; Hammon, K.; McCarthy, B.; Eisenhart, T.; Dempsey, J. A Practical Beginner’s Guide to Cyclic Voltammetry. J. Chem. Educ. 2017, 1–10. [Google Scholar] [CrossRef]
- Rashid, J.I.A.; Yusof, N.A. The strategies of DNA immobilization and hybridization detection mechanism in the construction of electrochemical DNA sensor: A review. Sens. BioSens. Res. 2017, 16, 19–31. [Google Scholar] [CrossRef]
- Focused Collection. Illustration of Transparent Blue Silhouette of Male Body with Colored Colon Cancer. 2020. Available online: https://focusedcollection.com/236856886/stock-photo-illustration-transparent-blue-silhouette-male.html (accessed on 16 December 2020).
- Dana-Farber Cancer Institute. What Is the Relationship Between Anemia and Cancer? 2020. Available online: https://blog.dana-farber.org/insight/2018/09/relationship-anemia-cancer/.ShutterstockctDNAimages (accessed on 27 November 2020).
- Shutterstock. ctDNA Images. 2020. Available online: https://www.shutterstock.com/search/ctdna (accessed on 27 November 2020).
- NCBI. 2020. Available online: https://www.ncbi.nlm.nih.gov/gene/3845 (accessed on 20 January 2020).
- Amplyus. Bluegel-Electrophoresis. 2017. Available online: https://www.minipcr.com/ (accessed on 7 October 2019).
- Thermofisher Scientific. Qubit 4 Fluorometer. 2020. Available online: https://www.thermofisher.com/uk/en/home/industrial/spectroscopy-elemental-isotope-analysis/molecular-spectroscopy/fluorometers/qubit/qubit-fluorometer.html (accessed on 14 October 2020).
- Hetemi, D.; Noël, V.; Pinson, J. Grafting of diazonium salts on surfaces: Application to biosensors. Biosensors 2020, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Chow, A.M.; Ganesh, H.V.S.; Ratnam, M.; Brown, I.R.; Kerman, K. Diazonium-modified screen-printed electrodes for immunosensing growth hormone in blood samples. Biosensors 2019, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Obaje, E.A.; Cummins, G.; Schulze, H.; Mahmood, S.; Desmulliez, M.P.Y.; Bachmann, T.T. Carbon screen-printed electrodes on ceramic substrates for label-free molecular detection of antibiotic resistance. J. Interdiscip. Nanomed. 2016, 1, 93–109. [Google Scholar] [CrossRef]
- Ocaña, C.; Hayat, A.; Mishra, R.K.; Vasilescu, A.; del Valle, M.; Marty, J.L. Label free aptasensor for Lysozyme detection: A comparison of the analytical performance of two aptamers. Bioelectrochemistry 2015, 105, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalupczok, S.; Kurzweil, P.; Hartmann, H.; Schell, C. The Redox Chemistry of Ruthenium Dioxide: A Cyclic Voltammetry Study—Review and Revision. Int. J. Electrochem. 2018, 2018, 1273768. [Google Scholar] [CrossRef]
- Loew, N.; Fitriana, M.; Hiraka, K.; Sode, K.; Tsugawa, W. Characterization of electron mediator preference of aerococcus viridans-derived lactate oxidase for use in disposable enzyme sensor strips. Sens. Mater. 2017, 29, 1703–1711. [Google Scholar] [CrossRef] [Green Version]
- Su, W.Y.; Wang, S.M.; Cheng, S.H. Electrochemically pretreated screen-printed carbon electrodes for the simultaneous determination of aminophenol isomers. J. Electroanal. Chem. 2011, 651, 166–172. [Google Scholar] [CrossRef]
- Manzanares-Palenzuela, C.L.; Fernandes, E.G.R.; Lobo-Castañón, M.J.; López-Ruiz, B.; Zucolotto, V. Impedance sensing of DNA hybridization onto nanostructured phthalocyanine-modified electrodes. Electrochim. Acta 2016, 221, 86–95. [Google Scholar] [CrossRef]
- Perakis, S.; Speicher, M.R. Emerging concepts in liquid biopsies. BMC Med. 2017, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, L.; Su, Y.; Yue, Z.; Xing, T. Plasma cell-free DNA quantification is highly correlated to tumor burden in children with neuroblastoma. Cancer Med. 2018, 3022–3030. [Google Scholar] [CrossRef] [PubMed]
- Reece, M.; Saluja, H.; Hollington, P.; Karapetis, C.S.; Vatandoust, S.; Young, G.P.; Symonds, E.L. The Use of Circulating Tumor DNA to Monitor and Predict Response to Treatment in Colorectal Cancer. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demuth, C.; Spindler, K.L.G.; Johansen, J.S.; Pallisgaard, N.; Nielsen, D.; Hogdall, E.; Vittrup, B.; Sorensen, B.S. Measuring KRAS Mutations in Circulating Tumor DNA by Droplet Digital PCR and Next-Generation Sequencing. Transl. Oncol. 2018, 11, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, F.; Pancas, R.; Magalha, E.; Bernardo, E.; Antunes, M.J. Diagnostic value of surgical lung biopsy: Comparison with clinical and radiological diagnosis. Eur. J. Cardio Thorac. Surg. 2008, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NHS. Biopsy. 2018. Available online: https://www.nhs.uk/conditions/biopsy/ (accessed on 18 July 2019).
- Chung, C.H.; Kim, J.H. One-step isothermal detection of multiple KRAS mutations by forming SNP specific hairpins on a gold nanoshell. Analyst 2018, 143, 3544–3548. [Google Scholar] [CrossRef]
- Attoye, B.; Pou, C.; Blair, E.; Rinaldi, C.; Thomson, F.; Baker, M.J.; Corrigan, D.K. Developing a Low-Cost, Simple-to-Use Electrochemical Sensor for the Detection of Circulating Tumour DNA in Human Fluids. Biosensors 2020, 10. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KRAS G13D Probe and Primer Sequences | |
| 23 Bases Wild-Type Hybridisation Probe | TGGAGCTGGTGGCGTAGGCAAGA |
| 23 Bases Mutant Hybridisation Probe | TGGAGCTGGTGACGTAGGCAAGA |
| Forward Primer (Wild-Type) | TGTGGTAGTTGGAGCTGGTG |
| Forward Primer (Mutant) | TGTGGTAGTTGGAGCTGATG |
| PCR Probe (Mutant) | TCTTGCCTACGCCACCAGCTCCA |
| Reverse Primer | TTGTGGACGAATATGATCCAACA |
| KRAS G12D Probe and Primer Sequences | |
| 23 bases Wild-type Hybridisation Probe | AGTTGGAGCTGGTGGCGTAGGCA |
| 23 bases Mutant Hybridisation Probe | AGTTGGAGCTGATGGCGTAGGCA |
| Forward Primer (Wild-type) | TGTGGTAGTTGGAGCTGGTG |
| Forward Primer (Mutant) | TGTGGTAGTTGGAGCTGATG |
| Reverse Primer | TTGTGGACGAATATGATCCAACA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attoye, B.; Baker, M.J.; Thomson, F.; Pou, C.; Corrigan, D.K. Optimisation of an Electrochemical DNA Sensor for Measuring KRAS G12D and G13D Point Mutations in Different Tumour Types. Biosensors 2021, 11, 42. https://doi.org/10.3390/bios11020042
Attoye B, Baker MJ, Thomson F, Pou C, Corrigan DK. Optimisation of an Electrochemical DNA Sensor for Measuring KRAS G12D and G13D Point Mutations in Different Tumour Types. Biosensors. 2021; 11(2):42. https://doi.org/10.3390/bios11020042
Chicago/Turabian StyleAttoye, Bukola, Matthew J. Baker, Fiona Thomson, Chantevy Pou, and Damion K. Corrigan. 2021. "Optimisation of an Electrochemical DNA Sensor for Measuring KRAS G12D and G13D Point Mutations in Different Tumour Types" Biosensors 11, no. 2: 42. https://doi.org/10.3390/bios11020042