Immobilization Techniques for Aptamers on Gold Electrodes for the Electrochemical Detection of Proteins: A Review


Abstract
:1. Introduction
1.1. Aptamers in Biosensing
1.2. Principle of Electrochemical Measurements
1.2.1. Redox Mediators
1.2.2. Cyclic Voltammetry
1.2.3. Electrochemical Impedance Spectroscopy
1.3. Formation of Thiol Monolayers on Gold Surfaces
2. Aptamer Immobilization via Direct Thiolation or Thiolated Short Linkers
2.1. Thiolated Aptamers
2.2. Short Linkers

2.3. Drawbacks of Mercaptohexanol
3. Antifouling Strategies
3.1. Serum Proteins
3.2. Thioaromatic Monolayers
3.3. Zwitterionic Peptides
4. Amplification Techniques
4.1. Improved Surface Area
4.1.1. Spherical and Non-Spherical Gold Nanoparticles
4.1.2. Spherical AuNPs on 11-amino-1-undecanethiol SAM
4.1.3. Dendrimer
4.2. Binding of the Redox Mediator
4.3. Linkage or Elongation of the Aptamers
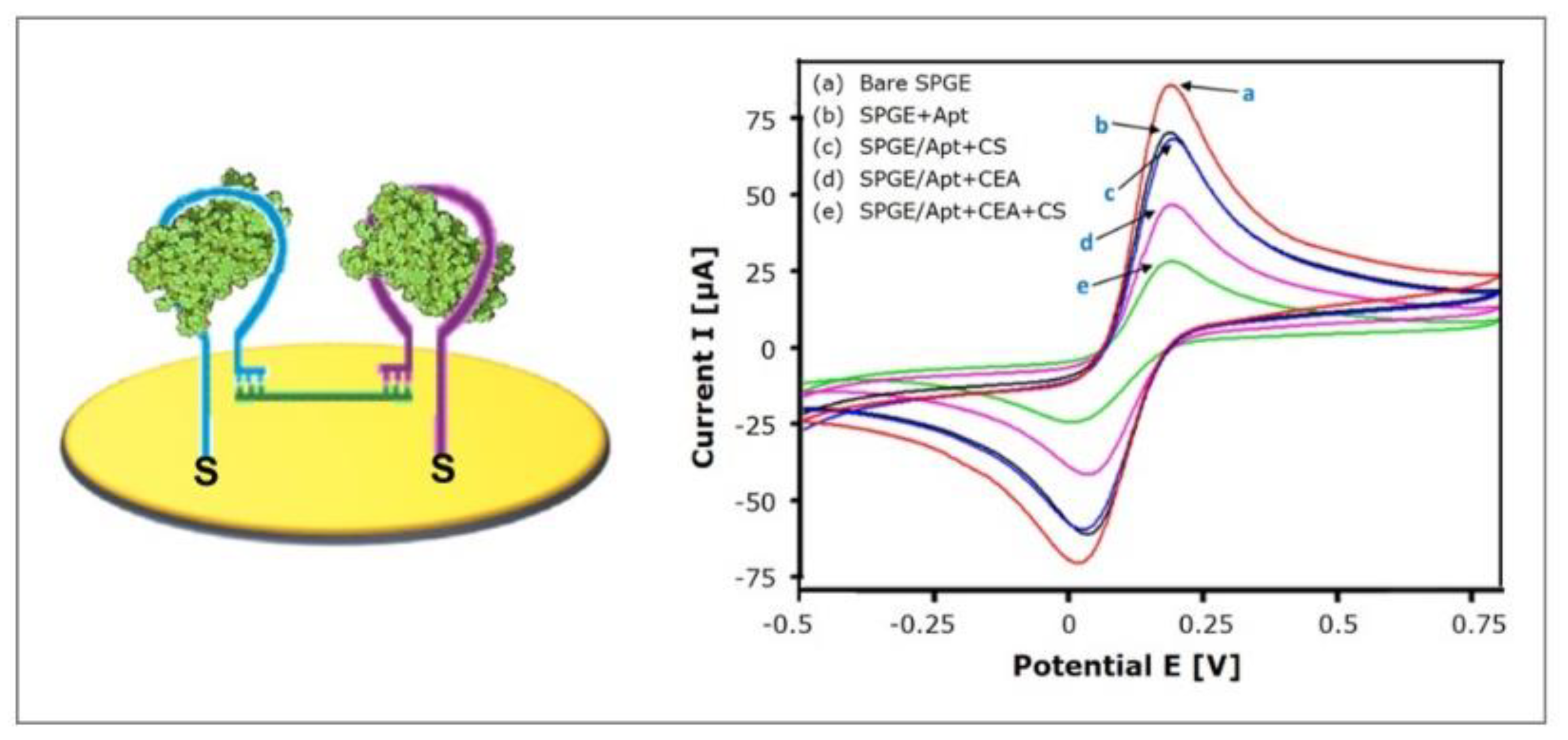
4.3.1. Target-Induced Bridge Assembly
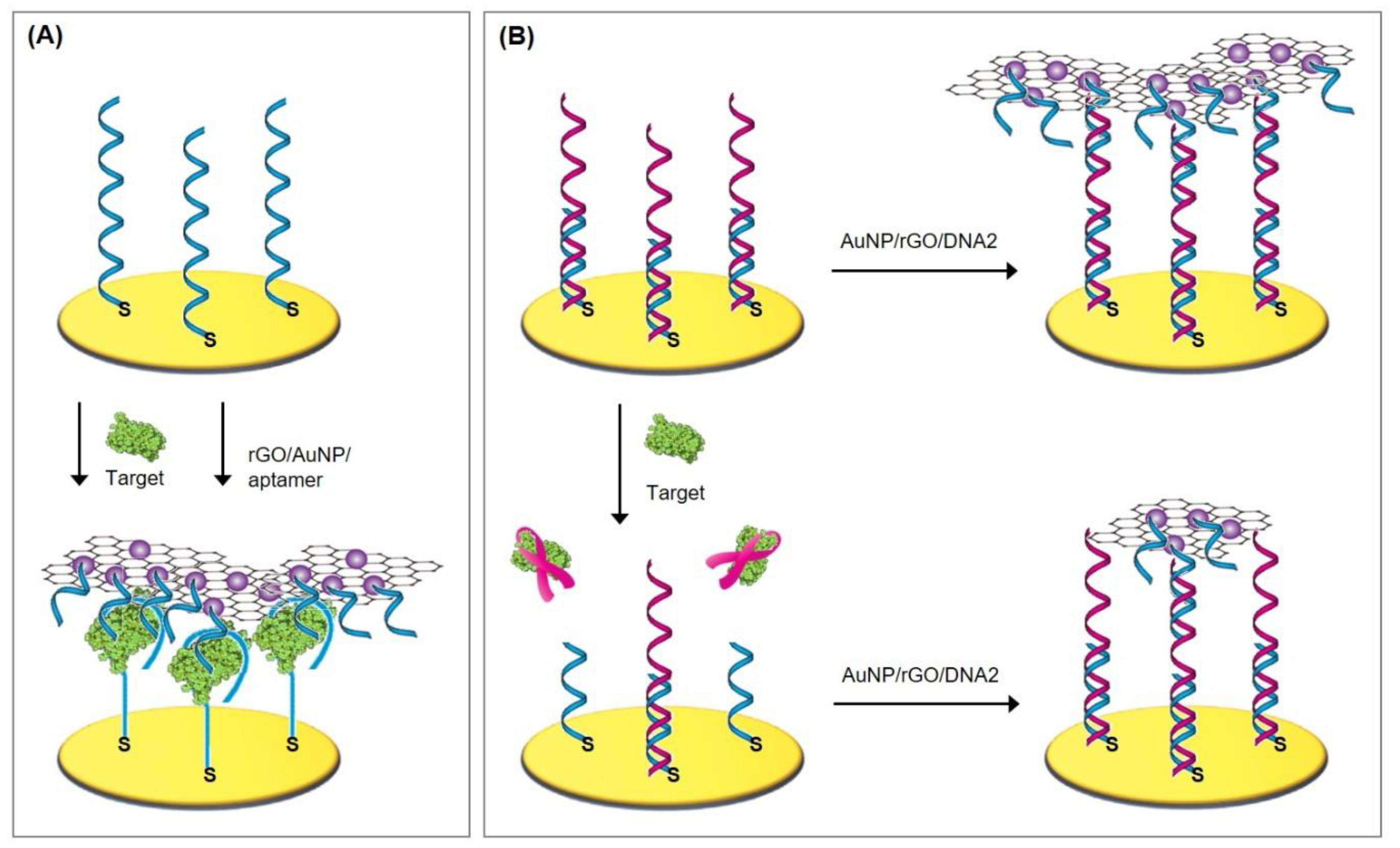
4.3.2. Amplification via Hybridization Chain Reaction
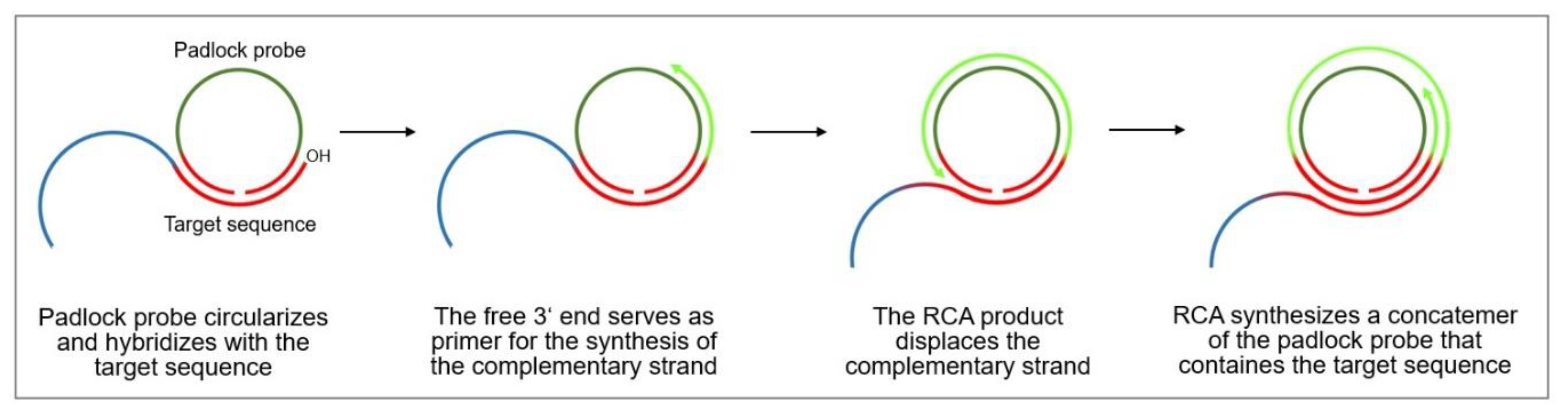
4.3.3. Rolling Circle Amplification
4.4. Graphene Nanosheets
5. Immobilization via Streptavidin/biotin Interactions, DNA Nanostructures, as well as Reduced Graphene Oxide and Pyrene
5.1. Immobilization via Streptavidin/avidin Interaction with Biotin
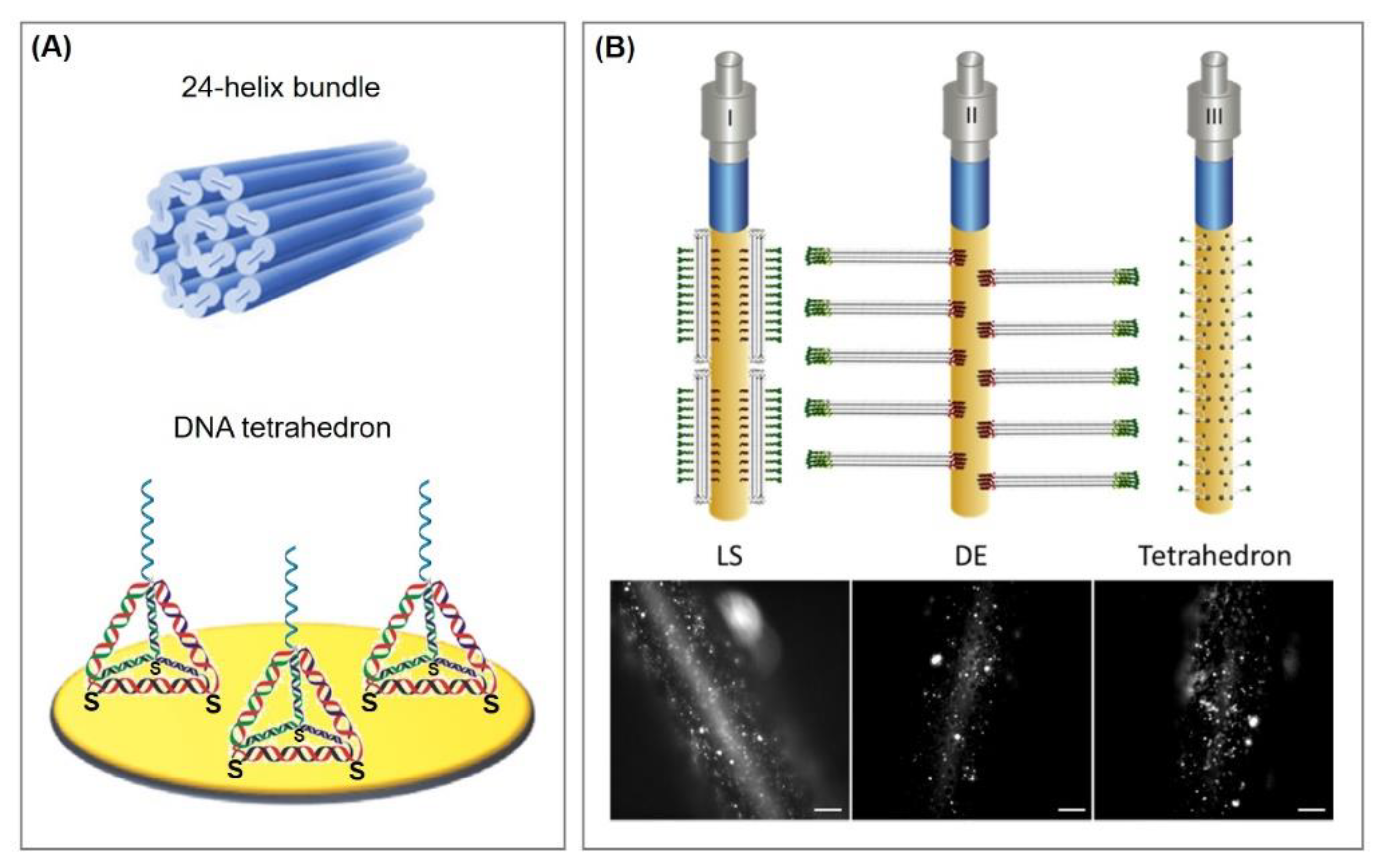
5.2. Immobilization via three-dimensional DNA nanostructures
5.3. Immobilization via Reduced Graphene Oxide, Pyrene, and Pyridine.
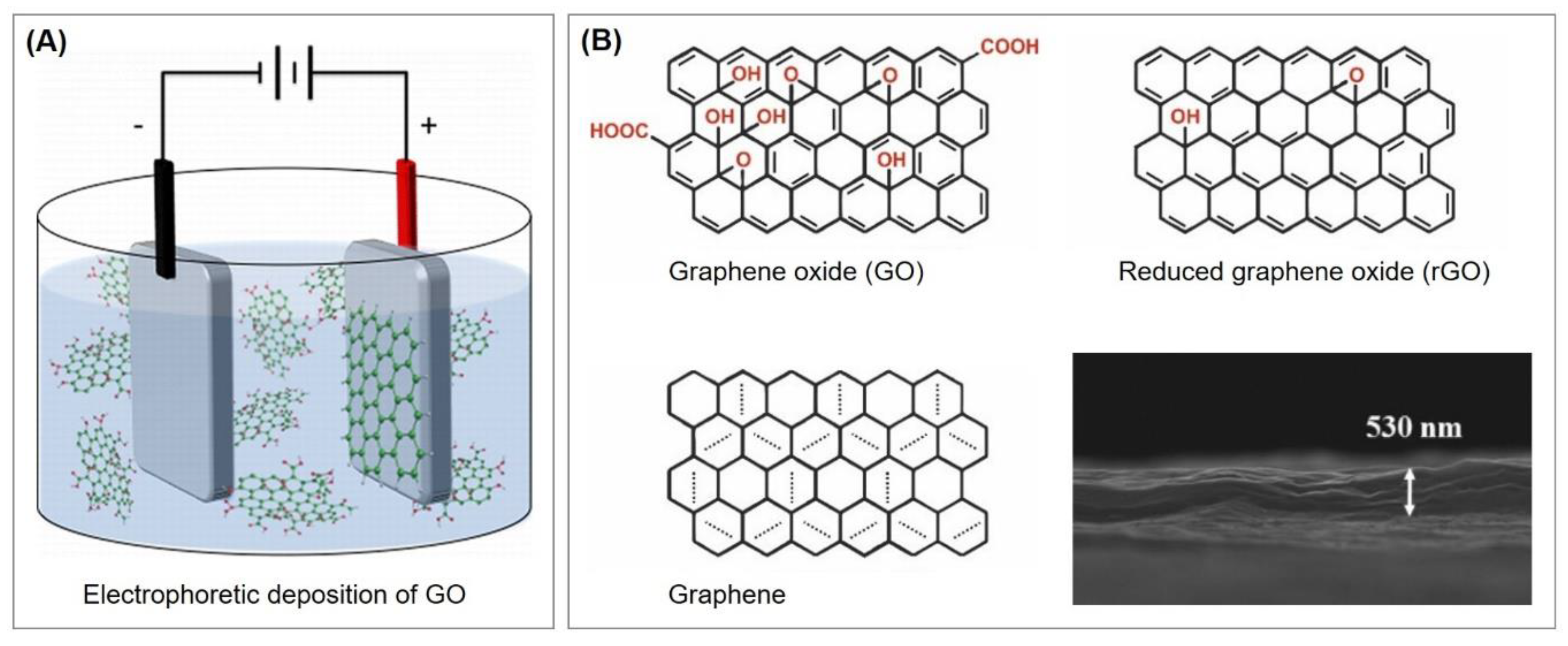
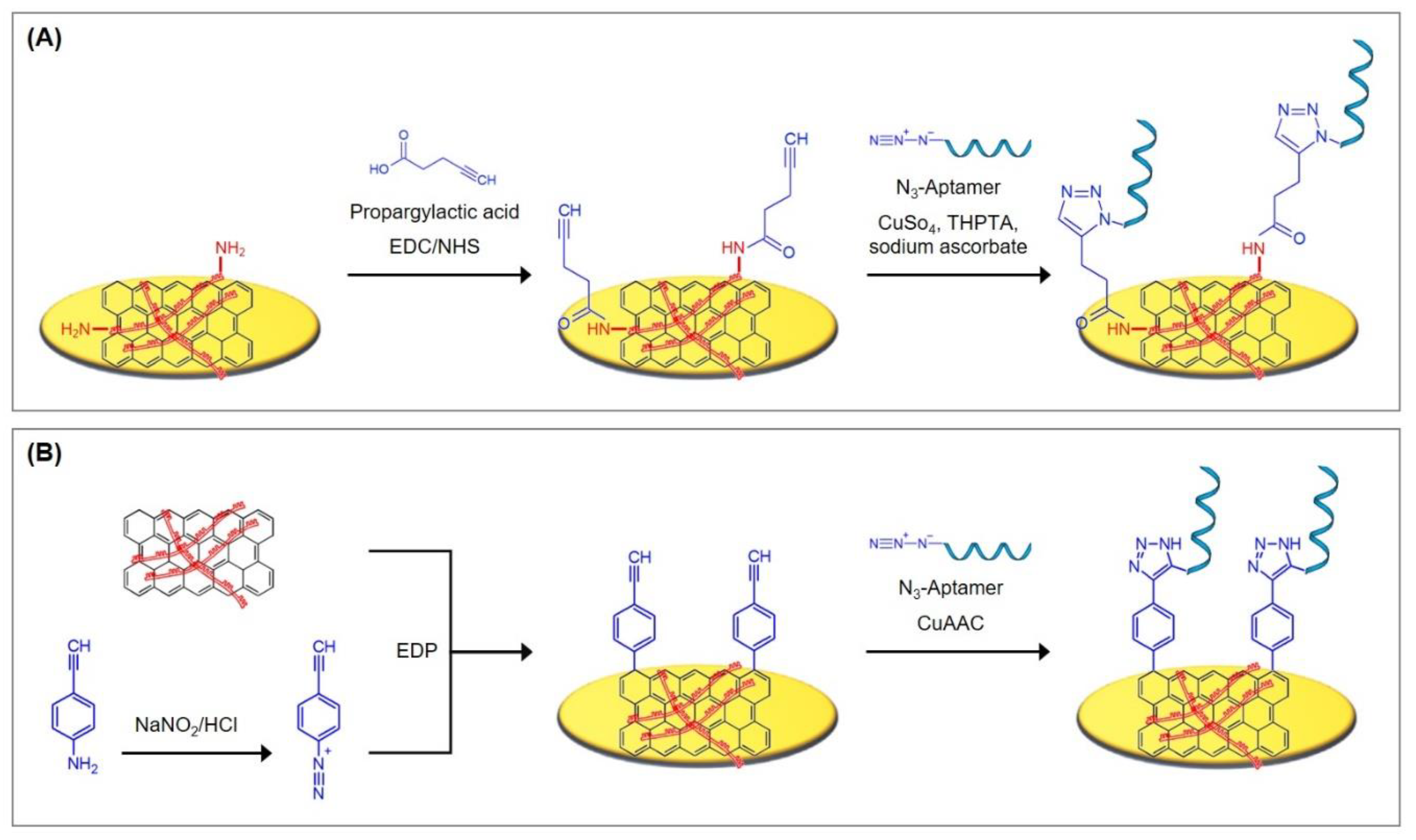
5.3.1. Reduced Graphene Oxide Deposits as Interface for Covalent Aptamer Immobilization
5.3.2. Aptamer Immobilization via Non-Covalent π–π Interactions of Graphene, Pyrene, and Porphyrin
6. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
| Protocol A1 by Ciu et al. [77]: Mixed monolayer of aptamer and zwitterionic peptide Reagents
| Protocol A2 by Peng et al. [110]: Signal amplification via graphene oxide and methylene blue binding Reagents
|
| Protocol A3 by Jolly et al. [78]: Spherical AuNPs on 11-amino-1-undecanethiol SAM Reagents
| Protocol A4 by Daems et al. [141]: 3D DNA tetrahedron as anchor for aptamer immobilization Reagents
|
| Protocol A5 by Negahdary et al. [80]: Electrodeposition of fern-leaf-like gold nanostructures with increased surface area for aptamer immobilization Reagents
For the bioassay, incubate with Aβ (linear range: 2 pg/mL–1.28 ng/mL, diluted in artificial CSF) for 10 min at 37 °C and rinse with deionized water. For regeneration, immerse in deionized water for 5 min at 95 °C to release bound Aβ. |
| Protocol A6 by Malvano et al. [42]: Cysteamine, glutaraldehyde, and PAMAM dendrimer scaffold Reagents
|
| Protocol A7 by Cao et al. [13]: Dual-signaling strategy of aptamer-labelling with ferrocene and the intercalation of RuHex Reagents
|
| Protocol A8 by Ding et al. [105]: Adamantane as surface anchor for aptamers and signal amplification via amplification chain reaction Reagents
|
| Protocol A9 by Meirinho et al. [120]: Immobilization of biotinylated aptamer via streptavidin Reagents
|
| Protocol A10 by Daems et al. [141]: Immobilization of 24-helix DNA bundle as anchor platform for aptamer immobilization Reagents
|
| Protocol A11 by Grabowska et al. [23]: Electrophoretic deposition of rGO/PEI nanocomposite and immobilization of azide-terminated aptamers onto PEI Reagents
|
| Protocol A12 by Wang et al. [24]: Electrophoretic deposition of rGO/PEI nanocomposite and immobilization of azide-terminated aptamers onto rGO Reagents
|
References
- Mukhopadhyay, R. Aptamers Are Ready for the Spotlight. Anal. Chem. 2005, 77, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lai, B.S.; Juhas, M. Recent Advances in Aptamer Discovery and Applications. Molecules 2019, 24, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, S.I.; Alshaer, W. Therapeutic aptamers in discovery, preclinical and clinical stages. Adv. Drug Deliv. Rev. 2018, 134, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J.J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2016, 16, 181–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, T. Adaptive Recognition by Nucleic Acid Aptamers. Science 2000, 287, 820–825. [Google Scholar] [CrossRef] [Green Version]
- Baird, G.S. Where are all the aptamers? Am. J. Clin. Pathol. 2010, 134, 529–531. [Google Scholar] [CrossRef] [Green Version]
- Sumitani, M.; Takagi, S.; Tanamura, Y.; Inoue, H. Oxygen indicator composed of an organic/inorganic hybrid compound of methylene blue, reductant, surfactant and saponite. Anal. Sci. 2004, 20, 1153–1157. [Google Scholar] [CrossRef] [Green Version]
- Ferapontova, E.E. Electrochemical Indicators for DNA Electroanalysis. Curr. Anal. Chem. 2011, 7, 51–62. [Google Scholar] [CrossRef]
- Elgrishi, N.; Rountree, K.J.; McCarthy, B.D.; Rountree, E.; Eisenhart, T.T.; Dempsey, J.L. A Practical Beginner’s Guide to Cyclic Voltammetry. J. Chem. Educ. 2017, 95, 197–206. [Google Scholar] [CrossRef]
- Bardini, L. EIS 101, an introduction to electrochemical spectroscopy. What was a website is now available as a self-contained PDF. Available online: https://www.researchgate.net/publication/280009629_EIS_101_an_introduction_to_electrochemical_spectroscopy_What_was_a_website_is_now_available_as_a_self-contained_PDF (accessed on 24 April 2020).
- Jiang, L.; Qian, J.; Yang, X.; Yan, Y.; Liu, Q.; Wang, K.; Wang, K. Amplified impedimetric aptasensor based on gold nanoparticles covalently bound graphene sheet for the picomolar detection of ochratoxin A. Anal. Chim. Acta 2014, 806, 128–135. [Google Scholar] [CrossRef]
- Taghdisi, S.M.; Danesh, N.M.; Nameghi, M.A.; Ramezani, M.; Alibolandi, M.; Hassanzadeh-Khayat, M.; Emrani, A.S.; Abnous, K. A Novel Electrochemical Aptasensor Based on Nontarget-Induced High Accumulation of Methylene Blue on the Surface of Electrode for Sensing of Alpha-Synuclein Oligomer. Biosens. Bioelectron. 2019, 123, 14–18. [Google Scholar] [CrossRef]
- Cao, X.; Xia, J.; Liu, H.; Zhang, F.; Wang, Z.; Lu, L. A new dual-signalling electrochemical aptasensor with the integration of “signal on/off” and “labeling/Label-free” strategies. Sens. Actuators B Chem. 2017, 239, 166–171. [Google Scholar] [CrossRef]
- Fan, T.; Du, Y.; Yao, Y.; Wu, J.; Meng, S.; Luo, J.; Zhang, X.; Yang, D.; Wang, C.; Qian, Y.; et al. Rolling circle amplification triggered poly adenine-gold nanoparticles production for label-free electrochemical detection of thrombin. Sens. Actuators B Chem. 2018, 266, 9–18. [Google Scholar] [CrossRef]
- Luderer, F.; Walschus, U. Immobilization of Oligonucleotides for Biochemical Sensing by Self-Assembled Monolayers: Thiol-Organic Bonding on Gold and Silanization on Silica Surfaces, Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Ulman, A. Formation and Structure of Self-Assembled Monolayers. Chem. Rev. 1996, 96, 1533–1554. [Google Scholar] [CrossRef] [PubMed]
- Doblhofer, K.; Figura, J.; Fuhrhop, J.H. Stability and electrochemical behavior of "self-assembled" adsorbates with terminal ionic groups. Langmuir 1992, 8, 1811–1816. [Google Scholar] [CrossRef]
- Vericat, C.; Vela, M.E.; Benitez, G.; Carro, P.; Salvarezza, R.C. Self-Assembled Monolayers of Thiols and Dithiols on Gold: New Challenges for a Well-Known System. Chem. Soc. Rev. 2010, 39, 1805–1834. [Google Scholar] [CrossRef]
- Kepley, L.J.; Crooks, R.M.; Ricco, A.J. Selective Surface Acoustic Wave-Based Organophosphonate Chemical Sensor Employing a Self-Assembled Composite Monolayer: A New Paradigm for Sensor Design. Anal. Chem. 1992, 64, 3191–3193. [Google Scholar] [CrossRef]
- Porter, M.D.; Bright, T.B.; Allara, D.L.; Chidsey, C.E.D. Spontaneously organized molecular assemblies. 4. Structural characterization of n-alkyl thiol monolayers on gold by optical ellipsometry, infrared spectroscopy, and electrochemistry. J. Am. Chem. Soc. 1987, 109, 3559–3568. [Google Scholar] [CrossRef]
- Chidsey, C.E.D.; LoIacono, D.N. Chemical functionality in self-assembled monolayers: Structural and electrochemical properties. Langmuir 1990, 6, 682–691. [Google Scholar] [CrossRef]
- Kong, N.; Liu, J.; Kong, Q.; Wang, R.; Barrow, C.J.; Yang, W. Graphene modified gold electrode via π–π stacking interaction for analysis of Cu2+ and Pb2+. Sens. Actuators B Chem. 2013, 178, 426–433. [Google Scholar] [CrossRef]
- Grabowska, I.; Sharma, N.; Vasilescu, A.; Iancu, M.; Badea, G.; Boukherroub, R.; Ogale, S.; Szunerits, S. Electrochemical Aptamer-Based Biosensors for the Detection of Cardiac Biomarkers. ACS Omega 2018, 3, 12010–12018. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Vasilescu, A.; Wang, Q.; Coffinier, Y.; Li, M.; Boukherroub, R.; Szunerits, S. Electrophoretic Approach for the Simultaneous Deposition and Functionalization of Reduced Graphene Oxide Nanosheets with Diazonium Compounds: Application for Lysozyme Sensing in Serum. ACS Appl. Mater. Interfaces 2017, 9, 12823–12831. [Google Scholar] [CrossRef]
- Leff, D.V.; Brandt, L.; Heath, J.R. Synthesis and Characterization of Hydrophobic, Organically-Soluble Gold Nanocrystals Functionalized with Primary Amines. Langmuir 1996, 12, 4723–4730. [Google Scholar] [CrossRef]
- Levicky, R.; Herne, T.M.; Tarlov, M.J.; Satija, S.K. Using Self-Assembly To Control the Structure of DNA Monolayers on Gold: A Neutron Reflectivity Study. J. Am. Chem. Soc. 1998, 120, 9787–9792. [Google Scholar] [CrossRef]
- Herne, T.M.; Tarlov, M.J. Characterization of DNA Probes Immobilized on Gold Surfaces. J. Am. Chem. Soc. 1997, 119, 8916–8920. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Gong, P.; Harbers, G.M.; Grainger, D.W.; Castner, D.G.; Gamble, L.J. Surface Coverage and Structure of Mixed DNA/Alkylthiolmonolayers on Gold: Characterization by Xps, Nexafs, Andfluorescence Intensity Measurements. Anal. Chem. 2006, 78, 3326–3334. [Google Scholar] [CrossRef] [Green Version]
- Keighley, S.D.; Li, P.; Estrela, P.; Migliorato, P. Optimization of DNA immobilization on gold electrodes for label-free detection by electrochemical impedance spectroscopy. Biosens. Bioelectron. 2008, 23, 1291–1297. [Google Scholar] [CrossRef]
- Xu, X.; Makaraviciute, A.; Kumar, S.; Wen, C.; Sjödin, M.; Abdurakhmanov, E.; Danielson, U.H.; Nyholm, L.; Zhang, Z. Structural Changes of Mercaptohexanol Self-Assembled Monolayers on Gold and Their Influence on Impedimetric Aptamer Sensors. Anal. Chem. 2019, 91, 14697–14704. [Google Scholar] [CrossRef]
- Bábelová, L.; Sohová, M.E.; Poturnayova, A.; Buríková, M.; Bizik, J.; Hianik, T. Label-free Electrochemical Aptasensor for Jurkat Cells Detection as a Potential Diagnostic Tool for Leukemia. Electroanalysis 2018, 30, 1487–1495. [Google Scholar] [CrossRef]
- Jarczewska, M.; Ziółkowski, R.; Gorski, L.; Malinowska, E. Application of RNA Aptamers as Recognition Layers for the Electrochemical Analysis of C-Reactive Protein. Electroanalysis 2017, 30, 658–664. [Google Scholar] [CrossRef]
- Salimian, R.; Kékedy-Nagy, L.; Ferapontova, E.E. Specific Picomolar Detection of a Breast Cancer Biomarker HER-2/neu Protein in Serum: Electrocatalytically Amplified Electroanalysis by the Aptamer/PEG-Modified Electrode. ChemElectroChem 2017, 4, 872–879. [Google Scholar] [CrossRef]
- Ahirwar, R.; Dalal, A.; Sharma, J.G.; Yadav, B.K.; Nahar, P.; Kumar, A.; Kumar, S. An aptasensor for rapid and sensitive detection of estrogen receptor alpha in human breast cancer. Biotechnol. Bioeng. 2018, 116, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.K.; Arya, S.; Estrela, P.; Goswami, P. Capacitive malaria aptasensor using Plasmodium falciparum glutamate dehydrogenase as target antigen in undiluted human serum. Biosens. Bioelectron. 2018, 117, 246–252. [Google Scholar] [CrossRef]
- Reich, P.; Stoltenburg, R.; Strehlitz, B.; Frense, D.; Beckmann, D. Development of An Impedimetric Aptasensor for the Detection of Staphylococcus aureus. Int. J. Mol. Sci. 2017, 18, 2484. [Google Scholar] [CrossRef] [Green Version]
- Ying, G.; Wang, M.; Yi, Y.; Chen, J.; Mei, J.; Zhang, Y.; Chen, S. Construction and application of an electrochemical biosensor based on an endotoxin aptamer. Biotechnol. Appl. Biochem. 2017, 65, 323–327. [Google Scholar] [CrossRef]
- Bart, J.; Tiggelaar, R.M.; Yang, M.; Schlautmann, S.; Zuilhof, H.; Gardeniers, H.J.G.E. Room-temperature intermediate layer bonding for microfluidic devices. Lab Chip 2009, 9, 3481–3488. [Google Scholar] [CrossRef]
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science; John Wiley & Sons: Hoboken, NJ, USA, 2002; Volume 1. [Google Scholar]
- Hermanson, G.T. Bioconjugate Techniques, 3 ed.; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Chakma, B.; Jain, P.; Singh, N.K.; Goswami, P. Development of Electrochemical Impedance Spectroscopy Based Malaria Aptasensor Using HRP-II as Target Biomarker. Electroanalysis 2018, 30, 1847–1854. [Google Scholar] [CrossRef]
- Malvano, F.; Albanese, D.; Pilloton, R.; Di Matteo, M. A new label-free impedimetric aptasensor for gluten detection. Food Control. 2017, 79, 200–206. [Google Scholar] [CrossRef]
- Arduini, F.; Guidone, S.; Amine, A.; Palleschi, G.; Moscone, D. Acetylcholinesterase biosensor based on self-assembled monolayer-modified gold-screen printed electrodes for organophosphorus insecticide detection. Sens. Actuators B Chem. 2013, 179, 201–208. [Google Scholar] [CrossRef]
- Xiao, Y.; Ju, H.-X.; Chen, H. Hydrogen peroxide sensor based on horseradish peroxidase-labeled Au colloids immobilized on gold electrode surface by cysteamine monolayer. Anal. Chim. Acta 1999, 391, 73–82. [Google Scholar] [CrossRef]
- Yorganci, E.; Akyilmaz, E. Alkaline Phosphatase Based Amperometric Biosensor Immobilized by Cysteamine-Glutaraldehyde Modified Self-Assembled Monolayer. Artif. Cells Blood Substit. Biotechnol. 2011, 39, 317–323. [Google Scholar] [CrossRef]
- Eissa, S.; Abdulkarim, H.; Dasouki, M.; Al Mousa, H.; Arnout, R.; Al Saud, B.; Rahman, A.A.; Zourob, M. Multiplexed detection of DOCK8, PGM3 and STAT3 proteins for the diagnosis of Hyper-Immunoglobulin E syndrome using gold nanoparticles-based immunosensor array platform. Biosens. Bioelectron. 2018, 117, 613–619. [Google Scholar] [CrossRef]
- Tsai, W.-C.; Lin, I.-C. Development of a piezoelectric immunosensor for the detection of alpha-fetoprotein. Sens. Actuators B Chem. 2005, 106, 455–460. [Google Scholar] [CrossRef]
- Mattos, A.; Freitas, T.; Silva, V.; Dutra, R.F. A dual quartz crystal microbalance for human cardiac troponin T in real time detection. Sens. Actuators B Chem. 2012, 161, 439–446. [Google Scholar] [CrossRef]
- Gaffar, S.; Nurmalasari, R.; Hartati, Y.W. Yohan Voltammetric DNA Biosensor using Gold Electrode Modified by Self Assembled Monolayer of Thiol for Detection of Mycobacterium Tuberculosis. Procedia Technol. 2017, 27, 74–80. [Google Scholar] [CrossRef]
- Nurmalasari, R.; Gaffar, S.; Hartati, Y.W. Yohan Label-Free Electrochemical DNA Biosensor for the Detection of Mycobacterium Tuberculosis Using Gold Electrode Modified by Self-Assembled Monolayer of Thiol. Procedia Chem. 2015, 17, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, Y.; Liu, S.; Li, J.; Yao, S. Monitoring the Self-Assembly of Chitosan/Glutaraldehyde/Cysteamine/Au-Colloid and the Binding of Human Serum Albumin with Hesperidin. Biomaterials 2004, 25, 5725–5733. [Google Scholar] [CrossRef]
- Sun, J.-J.; Xu, J.-J.; Fang, H.-Q.; Chen, H. Electrocatalytical oxidation of NADH with dopamine covalently bound to self-assembled cysteamine monolayers on a gold electrode. Bioelectrochem. Bioenerg. 1997, 44, 45–50. [Google Scholar] [CrossRef]
- Miranda-Castro, R.; Salcedo, R.S.; Suárez-Álvarez, B.; De-Los-Santos-Álvarez, N.; Miranda-Ordieres, A.J.; Lobo-Castañón, M.J. Thioaromatic DNA monolayers for target-amplification-free electrochemical sensing of environmental pathogenic bacteria. Biosens. Bioelectron. 2017, 92, 162–170. [Google Scholar] [CrossRef]
- Campuzano, S.; Pedrero, M.; Yáñez-Sedeño, P.; Pingarrón, J.M. Antifouling (Bio)materials for Electrochemical (Bio)sensing. Int. J. Mol. Sci. 2019, 20, 423. [Google Scholar] [CrossRef] [Green Version]
- Josephs, E.A.; Ye, T. A Single-Molecule View of Conformational Switching of DNA Tethered to a Gold Electrode. J. Am. Chem. Soc. 2012, 134, 10021–10030. [Google Scholar] [CrossRef]
- Lao, R.; Song, S.; Wu, H.; Wang, L.; Zhang, Z.; He, L.; Fan, C. Electrochemical Interrogation of DNA Monolayers on Gold Surfaces. Anal. Chem. 2005, 77, 6475–6480. [Google Scholar] [CrossRef]
- Yang, F.; Zuo, X.; Fan, C.; Zhang, X.-E. Biomacromolecular nanostructures-based interfacial engineering: From precise assembly to precision biosensing. Natl. Sci. Rev. 2018, 5, 740–755. [Google Scholar] [CrossRef] [Green Version]
- Boozer, C.; Chen, S.; Jiang, S. Controlling DNA Orientation on Mixed ssDNA/OEG SAMs. Langmuir 2006, 22, 4694–4698. [Google Scholar] [CrossRef]
- Jolly, P.; Formisano, N.; Tkac, J.; Kasak, P.; Frost, C.; Estrela, P. Label-free impedimetric aptasensor with antifouling surface chemistry: A prostate specific antigen case study. Sens. Actuators B Chem. 2015, 209, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Campuzano, S.; Kuralay, F.; Lobo-Castañón, M.J.; Bartošík, M.; Vyavahare, K.; Paleček, E.; Haake, D.A.; Wang, J. Ternary monolayers as DNA recognition interfaces for direct and sensitive electrochemical detection in untreated clinical samples. Biosens. Bioelectron. 2011, 26, 3577–3583. [Google Scholar] [CrossRef] [Green Version]
- Titoiu, A.M.; Porumb, R.; Fanjul-Bolado, P.; Epure, P.; Zamfir, M.; Vasilescu, A. Detection of Allergenic Lysozyme during Winemaking with an Electrochemical Aptasensor. Electroanalysis 2019, 31, 2262–2273. [Google Scholar] [CrossRef]
- Youn, H.; Her, J.; Mok, J.; Kil, B.; Kim, E.; Park, H.; Ban, C. A Novel Eosinophilia Diagnostics Using Label-free Impedimetric Aptasensor for Soluble Interleukin-5 Receptor Alpha. Electroanalysis 2018, 30, 2597–2603. [Google Scholar] [CrossRef]
- Hanssen, B.; Siraj, S.; Wong, D. Recent strategies to minimise fouling in electrochemical detection systems. Rev. Anal. Chem. 2016, 35, 1–28. [Google Scholar] [CrossRef]
- Miodek, A.; Regan, E.M.; Bhalla, N.; Hopkins, N.A.; Goodchild, S.A.; Estrela, P. Optimisation and Characterisation of Anti-Fouling Ternary SAM Layers for Impedance-Based Aptasensors. Sensors 2015, 15, 25015–25032. [Google Scholar] [CrossRef] [Green Version]
- Lei, T.; He, Q.-Y.; Wang, Y.-L.; Si, L.-S.; Chiu, J.-F. Heparin chromatography to deplete high-abundance proteins for serum proteomics. Clin. Chim. Acta 2008, 388, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Nanjappa, V.; Thomas, J.K.; Marimuthu, A.; Muthusamy, B.; Radhakrishnan, A.; Sharma, R.; Khan, A.A.; Balakrishnan, L.; Sahasrabuddhe, N.A.; Kumar, S.; et al. Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res. 2013, 42, D959–D965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawthalkar, S.M. Essentials of Clinical Pathology; JP Medical Ltd.: New Delhi, India, 2018. [Google Scholar]
- McClatchey, K.D. Clinical Laboratory Medicine; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2002. [Google Scholar]
- Adult and Children’s Health Encyclopedia. University of Rochester Medical Center. Available online: https://www.urmc.rochester.edu/encyclopedia.aspx (accessed on 24 April 2020).
- Sabatani, E.; Cohen-Boulakia, J.; Bruening, M.; Rubinstein, I. Thioaromatic monolayers on gold: A new family of self-assembling monolayers. Langmuir 1993, 9, 2974–2981. [Google Scholar] [CrossRef]
- Hayes, W.A.; Shannon, C. Electrochemistry of Surface-Confined Mixed Monolayers of 4-Aminothiophenol and Thiophenol on Au. Langmuir 1996, 12, 3688–3694. [Google Scholar] [CrossRef]
- Miranda-Castro, R.; De-Los-Santos-Álvarez, N.; Lobo-Castañón, M.J. Understanding the Factors Affecting the Analytical Performance of Sandwich-hybridization Genosensors on Gold Electrodes. Electroanalysis 2018, 30, 1229–1240. [Google Scholar] [CrossRef]
- Zheng, J.; Li, L.; Tsao, H.-K.; Sheng, Y.-J.; Chen, S.; Jiang, S. Strong Repulsive Forces between Protein and Oligo (Ethylene Glycol) Self-Assembled Monolayers: A Molecular Simulation Study. Biophys. J. 2005, 89, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zheng, J.; Li, L.; Jiang, S. Strong Resistance of Phosphorylcholine Self-Assembled Monolayers to Protein Adsorption: Insights into Nonfouling Properties of Zwitterionic Materials. J. Am. Chem. Soc. 2005, 127, 14473–14478. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, F.; Shen, W.; Cheng, G.; Zhao, J.; Peng, W.; Qu, J. Amino acid-based anti-fouling functionalization of silica nanoparticles using divinyl sulfone. Acta Biomater. 2016, 40, 273–281. [Google Scholar] [CrossRef]
- Chelmowski, R.; Köster, S.D.; Kerstan, A.; Prekelt, A.; Grunwald, C.; Winkler, T.; Metzler-Nolte, N.; Terfort, A.; Wöll, C. Peptide-Based SAMs that Resist the Adsorption of Proteins. J. Am. Chem. Soc. 2008, 130, 14952–14953. [Google Scholar] [CrossRef]
- Cui, M.; Wang, Y.; Jiao, M.; Jayachandran, S.; Wu, Y.; Fan, X.; Luo, X. Mixed Self-Assembled Aptamer and Newly Designed Zwitterionic Peptide as Antifouling Biosensing Interface for Electrochemical Detection of alpha-Fetoprotein. ACS Sens. 2017, 2, 490–494. [Google Scholar] [CrossRef]
- Jolly, P.; Zhurauski, P.; Hammond, J.L.; Miodek, A.; Liébana, S.; Bertok, T.; Tkac, J.; Estrela, P. Self-assembled gold nanoparticles for impedimetric and amperometric detection of a prostate cancer biomarker. Sens. Actuators B Chem. 2017, 251, 637–643. [Google Scholar] [CrossRef]
- Paunovic, M.; Schlesinger, M. Fundamentals of Electrochemical Deposition, Second Edition, 45th ed.; The Ecs Series of Texts and Monographs; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Negahdary, M.; Heli, H. An ultrasensitive electrochemical aptasensor for early diagnosis of Alzheimer’s disease, using a fern leaves-like gold nanostructure. Talanta 2019, 198, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Khashayar, P.; Amoabediny, G.; Larijani, B.; Hosseini, M.; Verplancke, R.; Schaubroeck, D.; de Keersmaecker, M.; Adriaens, A.; Vanfleteren, J. Characterization of Gold Nanoparticle Layer Deposited on Gold Electrode by Various Techniques. Biointerface Res. Appl. Chem. 2016, 6, 1380–1390. [Google Scholar]
- Katzur, V.; Eichler, M.; Deigele, E.; Stage, C.; Karageorgiev, P.; Geis-Gerstorfer, J.; Schmalz, G.; Ruhl, S.; Rupp, F.; Mueller, R. Surface-immobilized PAMAM-dendrimers modified with cationic or anionic terminal functions: Physicochemical surface properties and conformational changes after application of liquid interface stress. J. Colloid Interface Sci. 2012, 366, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Bewley, C.A.; Gronenborn, A.M.; Clore, G.M. Minor Groove-Binding Architectural Proteins: Structure, Function, and DNA Recognition. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 105–131. [Google Scholar] [CrossRef]
- Nakano, T. Π-Stacked Polymers and Molecules: Theory, Synthesis, and Properties; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Williams, L.D. Molecular Interactions (Noncovalent Interactions) and the Behaviors of Biological Macromolecules. Available online: https://ww2.chemistry.gatech.edu/~lw26/structure/molecular_interactions/mol_int.html (accessed on 24 April 2020).
- Tani, A.; Thomson, A.J.; Butt, J. Methylene blue as an electrochemical discriminator of single- and double-stranded oligonucleotides immobilised on gold substrates. Analyst 2001, 126, 1756–1759. [Google Scholar] [CrossRef]
- Boon, E.M.; Jackson, N.M.; Wightman, M.D.; Kelley, S.; Hill, M.G.; Barton, J.K. Intercalative Stacking: A Critical Feature of DNA Charge-Transport Electrochemistry. J. Phys. Chem. B 2003, 107, 11805–11812. [Google Scholar] [CrossRef] [Green Version]
- Hianik, T.; Wang, J. Electrochemical Aptasensors—Recent Achievements and Perspectives. Electroanalysis 2009, 21, 1223–1235. [Google Scholar] [CrossRef]
- Citartan, M.; Tang, T.-H. Recent developments of aptasensors expedient for point-of-care (POC) diagnostics. Talanta 2019, 199, 556–566. [Google Scholar] [CrossRef]
- Xiao, M.; Lai, W.; Man, T.; Chang, B.; Li, L.; Chandrasekaran, A.R.; Pei, H. Rationally Engineered Nucleic Acid Architectures for Biosensing Applications. Chem. Rev. 2019, 119, 11631–11717. [Google Scholar] [CrossRef]
- Sahin, O.; Ziaei, A.; Karaismailoğlu, E.; Taheri, N. The serum angiotensin converting enzyme and lysozyme levels in patients with ocular involvement of autoimmune and infectious diseases. BMC Ophthalmol. 2016, 16, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firkin, F. Diagnostic value of the serum lysozyme (Muramidase) level. Pathology 1971, 3, 76. [Google Scholar] [CrossRef]
- Foulds, P.G.; Diggle, P.; Mitchell, J.D.; Parker, A.; Hasegawa, M.; Masuda-Suzukake, M.; Mann, D.M.A.; Allsop, D. A Longitudinal Study on A-Synuclein in Blood Plasma as a Biomarker for Parkinson’s Disease. Sci. Rep. 2013, 3, 2540. [Google Scholar] [CrossRef]
- Taghdisi, S.M.; Danesh, N.M.; Ramezani, M.; Emrani, A.S.; Abnous, K. A Novel Electrochemical Aptasensor for Carcinoembryonic Antigen Detection Based on Target-induced Bridge Assembly. Electroanalysis 2018, 30, 1734–1739. [Google Scholar] [CrossRef]
- Dirks, R.M.; Pierce, N. Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. USA 2004, 101, 15275–15278. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Yang, Y.; Chen, F.; Liu, T.; Jin, J.; Pan, Y.; Miao, P. Electrochemical detection of arsenic contamination based on hybridization chain reaction and RecJf exonuclease-mediated amplification. Chem. Eng. J. 2018, 353, 305–310. [Google Scholar] [CrossRef]
- Bao, T.; Wen, W.; Zhang, X.; Xia, Q.; Wang, S. An exonuclease-assisted amplification electrochemical aptasensor for Hg2+ detection based on hybridization chain reaction. Biosens. Bioelectron. 2015, 70, 318–323. [Google Scholar] [CrossRef]
- Zhou, Q.; Lin, Y.; Lin, Y.; Wei, Q.; Chen, G.; Tang, D. In situ amplified electrochemical aptasensing for sensitive detection of adenosine triphosphate by coupling target-induced hybridization chain reaction with the assembly of silver nanotags. Talanta 2016, 146, 23–28. [Google Scholar] [CrossRef]
- Jia, L.-P.; Wang, L.; Ma, R.; Shang, L.; Zhang, W.; Xue, Q.; Wang, H. An Electrochemical Aptasensor for the Highly Sensitive Detection of 8-Hydroxy-2′-Deoxyguanosine Based on the Hybridization Chain Reaction. Talanta 2018, 179, 414–419. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, L.; Ning, G.; Wu, Y.; Wu, S.; Mao, S.; Liu, G.-Q. An electrochemical strategy for tetracycline detection coupled triple helix aptamer probe with catalyzed hairpin assembly signal amplification. Biosens. Bioelectron. 2019, 143, 111613. [Google Scholar] [CrossRef]
- Zeng, R.; Su, L.; Luo, Z.; Zhang, L.; Lu, M.; Tang, D. Ultrasensitive and label-free electrochemical aptasensor of kanamycin coupling with hybridization chain reaction and strand-displacement amplification. Anal. Chim. Acta 2018, 1038, 21–28. [Google Scholar] [CrossRef]
- Zhao, L.; Huang, Y.; Qi, X.; Yan, X.; Wang, S.; Liang, X. Nanotetrahedron-assisted electrochemical aptasensor with cooperatively-folding aptamer chimera for sensitive and selective detection of lysozyme in red wines. Anal. Chim. Acta 2019, 1095, 172–178. [Google Scholar] [CrossRef]
- Luo, C.; Lei, Y.; Yan, L.; Yu, T.; Li, Q.; Zhang, D.; Ding, S.; Ju, H. A Rapid and Sensitive Aptamer-Based Electrochemical Biosensor for Direct Detection of Escherichia Coli O111. Electroanalysis 2012, 24, 1186–1191. [Google Scholar] [CrossRef]
- Jia, L.-P.; Zhao, R.-N.; Wang, L.-J.; Ma, R.-N.; Zhang, W.; Shang, L.; Wang, H. Aptamer based electrochemical assay for protein kinase activity by coupling hybridization chain reaction. Biosens. Bioelectron. 2018, 117, 690–695. [Google Scholar] [CrossRef]
- Ding, S.; Gu, Z.; Yan, R.; Tang, Y.; Miao, P. A novel mode of DNA assembly at electrode and its application to protein quantification. Anal. Chim. Acta 2018, 1029, 24–29. [Google Scholar] [CrossRef]
- Snustad, P.; Simmons, M.J. Principles of Genetics, 7th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Hartl, D.L.; Jones, E.W. Genetics: Analysis of Genes and Genomes; Jones & Bartlett Learning: Burlington, MA, USA, 2009. [Google Scholar]
- Ali, M.M.; Li, F.; Zhang, Z.; Zhang, K.; Kang, N.-K.; Ankrum, J.A.; Le, X.C.; Zhao, W. Rolling circle amplification: A versatile tool for chemical biology, materials science and medicine. Chem. Soc. Rev. 2014, 43, 3324–3341. [Google Scholar] [CrossRef]
- Demidov, V.V. Rolling Circle Amplification (Rca): Toward New Clinical Diagnostics and Therapeutics; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Peng, H.; Hui, Y.; Ren, R.; Wang, B.; Song, S.; He, Y.; Zhang, F. A sensitive electrochemical aptasensor based on MB-anchored GO for the rapid detection of Cronobacter sakazakii. J. Solid State Electrochem. 2019, 23, 3391–3398. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, Z.; Zhai, Y.; Zhang, L.; Hong, W.; Zhang, Z.; Dong, S. Label-free aptamer biosensor for thrombin detection based on functionalized graphene nanocomposites. Talanta 2015, 141, 247–252. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, H.-Y.; Xing, C.; Guo, M.; Xu, F.; Wang, X.; Gruber, H.J.; Zhang, B.; Tang, J. Sodium Citrate: A Universal Reducing Agent for Reduction / Decoration of Graphene Oxide with Au Nanoparticles. Nano Res. 2011, 4, 599–611. [Google Scholar] [CrossRef]
- Gilje, S.; Han, S.; Wang, M.; Wang, K.L.; Kaner, R.B. A Chemical Route to Graphene for Device Applications. Nano Lett. 2007, 7, 3394–3398. [Google Scholar] [CrossRef]
- Kuang, H.; Chen, W.; Xu, D.; Xu, L.; Zhu, Y.; Liu, L.; Chu, H.; Peng, C.; Xu, C.; Zhu, S. Fabricated aptamer-based electrochemical “signal-off” sensor of ochratoxin A. Biosens. Bioelectron. 2010, 26, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Weber, P.; Ohlendorf, D.; Wendoloski, J.; Salemme, F. Structural origins of high-affinity biotin binding to streptavidin. Science 1989, 243, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Livnah, O.; Bayer, E.A.; Wilchek, M.; Sussman, J.L. Three-Dimensional Structures of Avidin and the Avidinbiotin Complex. Proc. Natl. Acad. Sci. USA 1993, 90, 5076–5080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.N. Avidin and Streptavidin. In Avidin-Biotin Technology; Wilchek, M., Bayer, E.A., Eds.; Academic Press: Cambridge, MA, USA, 1990; pp. 51–67. [Google Scholar]
- Bank, RCSB Protein Data. 3ry1 Wild-Type Core Streptavidin at Atomic Resolution. Available online: http://www.rcsb.org/structure/3RY1 (accessed on 30 March 2020).
- Meirinho, S.; Dias, L.; Peres, A.M.; Rodrigues, L.R. Development of an electrochemical RNA-aptasensor to detect human osteopontin. Biosens. Bioelectron. 2015, 71, 332–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meirinho, S.; Dias, L.; Peres, A.M.; Rodrigues, L.R. Electrochemical aptasensor for human osteopontin detection using a DNA aptamer selected by SELEX. Anal. Chim. Acta 2017, 987, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Heller, M.J. DNA Microarray Technology: Devices, Systems, and Applications. Annu. Rev. Biomed. Eng. 2002, 4, 129–153. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.L.S.; Chow, E.; Gooding, J.J. DNA Recognition Interfaces: The Influence of Interfacial Design on the Efficiency and Kinetics of Hybridization. Langmuir 2005, 21, 6957–6965. [Google Scholar] [CrossRef]
- Irving, D.; Gong, P.; Levicky, R. DNA Surface Hybridization: Comparison of Theory and Experiment. J. Phys. Chem. B 2010, 114, 7631–7640. [Google Scholar] [CrossRef]
- Wong, I.Y.; Melosh, N.A. An Electrostatic Model for DNA Surface Hybridization. Biophys. J. 2010, 98, 2954–2963. [Google Scholar] [CrossRef] [Green Version]
- Steel, A.; Levicky, R.; Herne, T.; Tarlov, M. Immobilization of nucleic acids at solid surfaces: Effect of oligonucleotide length on layer assembly. Biophys. J. 2000, 79, 975–981. [Google Scholar] [CrossRef] [Green Version]
- Pei, H.; Zuo, X.; Pan, D.; Shi, J.; Huang, Q.; Fan, C. Scaffolded biosensors with designed DNA nanostructures. NPG Asia Mater. 2013, 5, e51. [Google Scholar] [CrossRef]
- Pei, H.; Lu, N.; Wen, Y.; Song, S.; Liu, Y.; Yan, H.; Fan, C. A DNA Nanostructure-based Biomolecular Probe Carrier Platform for Electrochemical Biosensing. Adv. Mater. 2010, 22, 4754–4758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpini, G.; Lucarelli, F.; Marrazza, G.; Mascini, M. Oligonucleotide-modified screen-printed gold electrodes for enzyme-amplified sensing of nucleic acids. Biosens. Bioelectron. 2004, 20, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, Q.; Wang, L.; Zhang, G.-J.; Fan, C. Framework-Nucleic-Acid-Enabled Biosensor Development. ACS Sens. 2018, 3, 903–919. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, N.; Schlapak, R.; Kastner, M.; Armitage, D.; Chrzanowski, W.; Riener, J.; Hinterdorfer, P.; Ebner, A.; Howorka, S. A DNA Nanostructure for the Functional Assembly of Chemical Groups with Tunable Stoichiometry and Defined Nanoscale Geometry. Angew. Chem. Int. Ed. Engl. 2009, 48, 525–527. [Google Scholar] [CrossRef]
- Wen, Y.; Liu, G.; Pei, H.; Li, L.; Xu, Q.; Liang, W.; Li, Y.; Xu, L.; Ren, S.; Fan, C. DNA nanostructure-based ultrasensitive electrochemical microRNA biosensor. Methods 2013, 64, 276–282. [Google Scholar] [CrossRef]
- Schlapak, R.; Danzberger, J.; Armitage, D.; Morgan, D.; Ebner, A.; Hinterdorfer, P.; Pollheimer, P.; Gruber, H.J.; Schäffler, F.; Howorka, S. Nanoscale DNA Tetrahedra Improve Biomolecular Recognition on Patterned Surfaces. Small 2011, 8, 89–97. [Google Scholar] [CrossRef]
- Madsen, M.; Gothelf, K.V. Chemistries for DNA Nanotechnology. Chem. Rev. 2019, 119, 6384–6458. [Google Scholar] [CrossRef]
- Bu, N.-N.; Tang, C.-X.; He, X.-W.; Yin, X.-B. Tetrahedron-structured DNA and functional oligonucleotide for construction of an electrochemical DNA-based biosensor. Chem. Commun. 2011, 47, 7689. [Google Scholar] [CrossRef]
- Wen, Y.; Pei, H.; Wan, Y.; Su, Y.; Huang, Q.; Song, S.; Fan, C. DNA Nanostructure-Decorated Surfaces for Enhanced Aptamer-Target Binding and Electrochemical Cocaine Sensors. Anal. Chem. 2011, 83, 7418–7423. [Google Scholar] [CrossRef]
- Ge, Z.; Lin, M.; Wang, P.; Pei, H.; Yan, J.; Shi, J.; Huang, Q.; He, D.; Fan, C.; Zuo, X. Hybridization Chain Reaction Amplification of MicroRNA Detection with a Tetrahedral DNA Nanostructure-Based Electrochemical Biosensor. Anal. Chem. 2014, 86, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Wen, Y.; Li, L.; Pei, H.; Liu, G.; Song, H.; Zuo, X.; Fan, C.; Huang, Q. Target-Responsive, DNA Nanostructure-Based E-DNA Sensor for microRNA Analysis. Anal. Chem. 2014, 86, 2285–2288. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, L.; Wan, S.; Cansiz, S.; Cui, C.; Liu, Y.; Cai, R.; Hong, C.-Y.; Teng, I.-T.; Shi, M.; et al. Aptasensor with Expanded Nucleotide Using DNA Nanotetrahedra for Electrochemical Detection of Cancerous Exosomes. ACS Nano 2017, 11, 3943–3949. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Lin, M.; Song, P.; Chen, X.; Chao, J.; Wang, L.; Huang, Q.; Huang, W.; Fan, C.; Zuo, X. Multivalent Capture and Detection of Cancer Cells with DNA Nanostructured Biosensors and Multibranched Hybridization Chain Reaction Amplification. Anal. Chem. 2014, 86, 7843–7848. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Wang, J.; Zhou, G.; Wang, J.; Wu, N.; Lu, J.; Gao, J.; Chen, X.; Shi, J.; Zuo, X.; et al. Programmable Engineering of a Biosensing Interface with Tetrahedral DNA Nanostructures for Ultrasensitive DNA Detection. Angew. Chem. Int. Ed. Engl. 2015, 54, 2151–2155. [Google Scholar] [CrossRef]
- Daems, D.; Pfeifer, W.; Rutten, I.; Saccà, B.; Spasic, D.; Lammertyn, J. Three-Dimensional DNA Origami as Programmable Anchoring Points for Bioreceptors in Fiber Optic Surface Plasmon Resonance Biosensing. ACS Appl. Mater. Interfaces 2018, 10, 23539–23547. [Google Scholar] [CrossRef]
- Pfeifer, W.; Lill, P.; Gatsogiannis, C.; Saccà, B. Hierarchical Assembly of DNA Filaments with Designer Elastic Properties. ACS Nano 2017, 12, 44–55. [Google Scholar] [CrossRef]
- Tadyszak, K.; Wychowaniec, J.K.; Litowczenko, J. Biomedical Applications of Graphene-Based Structures. Nanomaterials 2018, 8, 944. [Google Scholar] [CrossRef] [Green Version]
- Mishyn, V.; Aspermair, P.; Leroux, Y.; Happy, H.; Knoll, W.; Boukherroub, R.; Szunerits, S. “Click” Chemistry on Gold Electrodes Modified with Reduced Graphene Oxide by Electrophoretic Deposition. Surfaces 2019, 2, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, P.; Niedziolka-Jonsson, J.; Leśniewski, A.; Wang, Q.; Li, M.; Boukherroub, R.; Szunerits, S. Preparation of reduced graphene oxide–Ni(OH)2 composites by electrophoretic deposition: Application for non-enzymatic glucose sensing. J. Mater. Chem. A 2014, 2, 5525–5533. [Google Scholar] [CrossRef]
- Maaoui, H.; Singh, S.; Teodorescu, F.; Coffinier, Y.; Barras, A.; Chtourou, R.; Kurungot, S.; Szunerits, S.; Boukherroub, R. Copper oxide supported on three-dimensional ammonia-doped porous reduced graphene oxide prepared through electrophoretic deposition for non-enzymatic glucose sensing. Electrochim. Acta 2017, 224, 346–354. [Google Scholar] [CrossRef]
- Shen, B.; Hong, H.; Chen, S.; Chen, X.; Zhang, Z. Cathodic electrophoretic deposition of magnesium nitrate modified graphene coating as a macro-scale solid lubricant. Carbon 2019, 145, 297–310. [Google Scholar] [CrossRef]
- Jijie, R.; Kahlouche, K.; Barras, A.; Yamakawa, N.; Bouckaert, J.; Gharbi, T.; Szunerits, S.; Boukherroub, R. Reduced graphene oxide/polyethylenimine based immunosensor for the selective and sensitive electrochemical detection of uropathogenic Escherichia coli. Sens. Actuators B Chem. 2018, 260, 255–263. [Google Scholar] [CrossRef]
- Hwang, M.-J.; Kim, M.-G.; Kim, S.; Kim, Y.C.; Seo, H.W.; Cho, J.K.; Park, I.-K.; Suhr, J.; Moon, H.; Koo, J.C.; et al. Cathodic electrophoretic deposition (EPD) of phenylenediamine-modified graphene oxide (GO) for anti-corrosion protection of metal surfaces. Carbon 2019, 142, 68–77. [Google Scholar] [CrossRef]
- Ghazvini, A.S.; Taheri-Nassaj, E.; Raissi, B.; Riahifar, R.; Yaghmaee, M.S. Co-deposition of Co3O4 and graphene via electrophoretic technique. Mater. Lett. 2018, 213, 75–78. [Google Scholar] [CrossRef]
- Fraczek-Szczypta, A.; Jantas, D.; Ciepiela, F.; Grzonka, J. Graphene oxide-conductive polymer nanocomposite coatings obtained by the EPD method as substrates for neurite outgrowth. Diam. Relat. Mater. 2020, 102, 107663. [Google Scholar] [CrossRef]
- Jafari, Y.; Ghoreishi, S.M.; Shabani-Nooshabadi, M. Polyaniline/Graphene nanocomposite coatings on copper: Electropolymerization, characterization, and evaluation of corrosion protection performance. Synth. Met. 2016, 217, 220–230. [Google Scholar] [CrossRef]
- Behzadi, M.; Mirzaei, M. Poly(O-Anisidine)/Graphene Oxide Nanosheets Composite as a Coating for the Headspace Solid-Phase Microextraction of Benzene, Toluene, Ethylbenzene and Xylenes. J. Chromatogr. A 2016, 1443, 35–42. [Google Scholar] [CrossRef]
- Tang, X.; Raskin, J.-P.; Kryvutsa, N.; Hermans, S.; Slobodian, O.; Nazarov, A.N.; Debliquy, M. An ammonia sensor composed of polypyrrole synthesized on reduced graphene oxide by electropolymerization. Sens. Actuators B Chem. 2020, 305, 127423. [Google Scholar] [CrossRef]
- Ordikhani, F.; Farani, M.R.; Dehghani, M.; Tamjid, E.; Simchi, A. Physicochemical and biological properties of electrodeposited graphene oxide/chitosan films with drug-eluting capacity. Carbon 2015, 84, 91–102. [Google Scholar] [CrossRef]
- Ma, Y.; Han, J.; Wang, M.; Chen, X.; Jia, S. Electrophoretic deposition of graphene-based materials: A review of materials and their applications. J. Materiomics 2018, 4, 108–120. [Google Scholar] [CrossRef]
- Besra, L.; Liu, M. A review on fundamentals and applications of electrophoretic deposition (EPD). Prog. Mater. Sci. 2007, 52, 1–61. [Google Scholar] [CrossRef]
- Kasprzak, A.; Zuchowska, A.; Poplawska, M. Functionalization of graphene: Does the organic chemistry matter? Beilstein J. Org. Chem. 2018, 14, 2018–2026. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Valdez, A.; Shaffer, M.S.P.; Boccaccini, A. Applications of Graphene Electrophoretic Deposition. A Review. J. Phys. Chem. B 2012, 117, 1502–1515. [Google Scholar] [CrossRef]
- Wang, M.; Duong, L.D.; Oh, J.-S.; Mai, N.T.; Kim, S.; Hong, S.; Hwang, T.; Lee, Y.; Nam, J.-D. Large-Area, Conductive and Flexible Reduced Graphene Oxide (RGO) Membrane Fabricated by Electrophoretic Deposition (EPD). ACS Appl. Mater. Interfaces 2014, 6, 1747–1753. [Google Scholar] [CrossRef]
- Singh, M.; Holzinger, M.; Tabrizian, M.; Winters, S.; Berner, N.; Cosnier, S.; Duesberg, G.S. Noncovalently Functionalized Monolayer Graphene for Sensitivity Enhancement of Surface Plasmon Resonance Immunosensors. J. Am. Chem. Soc. 2015, 137, 2800–2803. [Google Scholar] [CrossRef] [PubMed]
- Chekin, F.; Vasilescu, A.; Jijie, R.; Singh, S.; Kurungot, S.; Iancu, M.; Badea, G.; Boukherroub, R.; Szunerits, S. Sensitive electrochemical detection of cardiac troponin I in serum and saliva by nitrogen-doped porous reduced graphene oxide electrode. Sens. Actuators B Chem. 2018, 262, 180–187. [Google Scholar] [CrossRef]
- Liu, D.; Zeng, Y.; Zhou, G.; Lu, X.; Miao, D.; Yang, Y.; Zhai, Y.; Zhang, J.; Zhang, Z.; Wang, H.; et al. Fluorometric determination of cardiac myoglobin based on energy transfer from a pyrene-labeled aptamer to graphene oxide. Microchim. Acta 2019, 186, 287. [Google Scholar] [CrossRef]
- Wu, G.; Dai, Z.; Tang, X.; Lin, Z.; Lo, P.K.; Meyyappan, M.; Lai, K.W.C. Graphene Field-Effect Transistors for the Sensitive and Selective Detection of Escherichia coli Using Pyrene-Tagged DNA Aptamer. Adv. Healthc. Mater. 2017, 6, 1700736. [Google Scholar] [CrossRef]
- Halouane, F.; Jijie, R.; Meziane, D.; Li, C.; Singh, S.K.; Bouckaert, J.; Jurazek, J.; Kurungot, S.; Barras, A.; Li, M.; et al. Selective isolation and eradication of E. coli associated with urinary tract infections using anti-fimbrial modified magnetic reduced graphene oxide nanoheaters. J. Mater. Chem. B 2017, 5, 8133–8142. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Pagneux, Q.; Larroulet, I.; Serrano, A.Y.; Pesquera, A.; Zurutuza, A.; Mandler, D.; Boukherroub, R.; Szunerits, S. Label-free femtomolar cancer biomarker detection in human serum using graphene-coated surface plasmon resonance chips. Biosens. Bioelectron. 2017, 89, 606–611. [Google Scholar] [CrossRef]
- Liu, J.; Bibari, O.; Mailley, P.; Dijon, J.; Rouvière, E.; Sauter-Starace, F.; Caillat, P.; Vinet, F.; Marchand, G. Stable non-covalent functionalisation of multi-walled carbon nanotubes by pyrene–polyethylene glycol through π–π stacking. New J. Chem. 2009, 33, 1017–1024. [Google Scholar] [CrossRef]
- Wang, Y.; Sauriat-Dorizon, H.; Korri-Youssoufi, H. Direct electrochemical DNA biosensor based on reduced graphene oxide and metalloporphyrin nanocomposite. Sens. Actuators B Chem. 2017, 251, 40–48. [Google Scholar] [CrossRef]
- Cao, Z.; Jia, Y.; Zhu, B. BNP and NT-proBNP as Diagnostic Biomarkers for Cardiac Dysfunction in Both Clinical and Forensic Medicine. Int. J. Mol. Sci. 2019, 20, 1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lydon, B.R.; Germann, A.; Yang, J.Y. Chemical modification of gold electrodes via non-covalent interactions. Inorg. Chem. Front. 2016, 3, 836–841. [Google Scholar] [CrossRef]
- Barras, A.; Szunerits, S.; Marcon, L.; Monfilliette-Dupont, N.; Boukherroub, R. Functionalization of Diamond Nanoparticles Using “Click” Chemistry. Langmuir 2010, 26, 13168–13172. [Google Scholar] [CrossRef] [PubMed]
- Aretz, J.; Boar, R.B.; Daugner, R.; Dürr, H.; Fischer, M. Houben-Weyl Methods of Organic Chemistry, 4th ed.; Photochemistry I ed. Vol. IV/5a; Georg Thieme Verlag: New York, NY, USA, 2014. [Google Scholar]
- Zhang, H.; Han, Y.; Guo, Y.; Dong, C. Porphyrin functionalized graphene nanosheets-based electrochemical aptasensor for label-free ATP detection. J. Mater. Chem. 2012, 22, 23900. [Google Scholar] [CrossRef]
- Zhang, H.; Shuang, S.; Sun, L.; Chen, A.; Qin, Y.; Dong, C. Label-free aptasensor for thrombin using a glassy carbon electrode modified with a graphene-porphyrin composite. Microchim. Acta 2013, 181, 189–196. [Google Scholar] [CrossRef]
- Hanna, C.M.; Sanborn, C.D.; Ardo, S.; Yang, J.Y. Interfacial Electron Transfer of Ferrocene Immobilized onto Indium Tin Oxide through Covalent and Noncovalent Interactions. ACS Appl. Mater. Interfaces 2018, 10, 13211–13217. [Google Scholar] [CrossRef]
- Saby, C.; Ortiz, B.; Champagne, G.Y.; Belanger, D. Electrochemical Modification of Glassy Carbon Electrode Using Aromatic Diazonium Salts. 1. Blocking Effect of 4-Nitrophenyl and 4-Carboxyphenyl Groups. Langmuir 1997, 13, 6805–6813. [Google Scholar] [CrossRef]
- Liu, J.; Tao, L.; Yang, W.; Li, D.; Boyer, C.; Wuhrer, R.; Braet, F.; Davis, T.P. Synthesis, Characterization, and Multilayer Assembly of pH Sensitive Graphene−Polymer Nanocomposites. Langmuir 2010, 26, 10068–10075. [Google Scholar] [CrossRef] [PubMed]
- Kadish, K.; Guilard, R.; Smith, K.M. The Porphyrin Handbook: Phthalocyanines: Spectroscopic and Electrochemical Characterization; Academic Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Miao, P.; Tang, Y.; Mao, Z.; Liu, Y. Adamantane Derivatives Functionalized Gold Nanoparticles for Colorimetric Detection of MiRNA. Part. Part. Syst. Charact. 2017, 34, 1600405. [Google Scholar] [CrossRef]
- Stahl, E.; Martin, T.G.; Praetorius, F.; Dietz, H. Facile and Scalable Preparation of Pure and Dense DNA Origami Solutions. Angew. Chem. Int. Ed. Engl. 2014, 53, 12735–12740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Aptamer Modifi-cation | Fabrication Procedure | Method | LOD | Linear Range | Ref |
|---|---|---|---|---|---|---|
| Protein tyrosine kinase-7 (PRK7) on leukemic Jurkat cells | 5′-thiol | Co-immobilization with MCH | EIS | 105 cells/mL | 50–500.000 cells/mL | [31] |
| C-reactive protein (CRP) | 5′-thiol-C6 | Backfilling with MCH | CV | 1 pM | 1–100 pM | [32] |
| HER-2/neu breast cancer biomarker | 5′-thiol-C6 | Backfilling with SH-C11-(EG)2-OH | CV | 1 pM | 1 pM–10 nM | [33] |
| Estrogen receptor alpha (ERα) | 5′-thiol-C6 | Only aptamer | DPV | 15 fM | 15 fM–15 nM | [34] |
| Plasmodium falciparum glutamate dehydro-genase (PfGDH) | 5‘-thiol-C6 | Co-immobilization and backfilling with MCH | EIS | 430 fM | 100 fM–100 nM | [35] |
| Protein A on Staphylococcus aureus | 3′-thiol-C6 | Co-immobilization and backfilling with MCH | EIS | 10 CFU/mL | N/A | [36] |
| Protein | Serum Concentration | Protein | Serum Concentration |
|---|---|---|---|
| Albumin | 35–50 mg/mL | Immunoglobulins | |
| Globulin | 18–35 mg/mL | ● IgA | ● 0.7–4 mg/mL |
| Fibrinogen | 2–4 mg/mL | ● IgE | ● 0.03–4 mg/mL |
| Haptoglobin | 0.3–2 mg/mL | ● IgG | ● 7–16 mg/mL |
| Serum cholesterol | Total < 2 mg/mL | ● IgM | ● 0.5–2.5 mg/mL |
| ● LDL | ● < 1.3 mg/mL | ● IgD | ● 0–0.08 mg/mL |
| ● HDL | ● 0.45 mg/mL | Transferrin | 0.17–0.37 mg/mL |
| ● Triglyceride | ● < 1.5 mg/mL | Hemoglobin | ≤0.05 mg/mL |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oberhaus, F.V.; Frense, D.; Beckmann, D. Immobilization Techniques for Aptamers on Gold Electrodes for the Electrochemical Detection of Proteins: A Review. Biosensors 2020, 10, 45. https://doi.org/10.3390/bios10050045
Oberhaus FV, Frense D, Beckmann D. Immobilization Techniques for Aptamers on Gold Electrodes for the Electrochemical Detection of Proteins: A Review. Biosensors. 2020; 10(5):45. https://doi.org/10.3390/bios10050045
Chicago/Turabian StyleOberhaus, Franziska V., Dieter Frense, and Dieter Beckmann. 2020. "Immobilization Techniques for Aptamers on Gold Electrodes for the Electrochemical Detection of Proteins: A Review" Biosensors 10, no. 5: 45. https://doi.org/10.3390/bios10050045