Shape-Stabilized Phase Change Materials for Solar Energy Storage: MgO and Mg(OH)2 Mixed with Polyethylene Glycol


Abstract
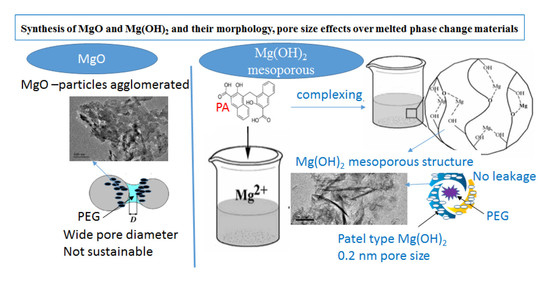
:
1. Introduction
2. Methodology
2.1. Chemicals
2.2. Hydrothermal Synthesis
2.3. Preparation of the Shape-Stabilized Composite PCM
2.4. Characterization
3. Results and Discussion
3.1. Characteristics of the Synthesized Products
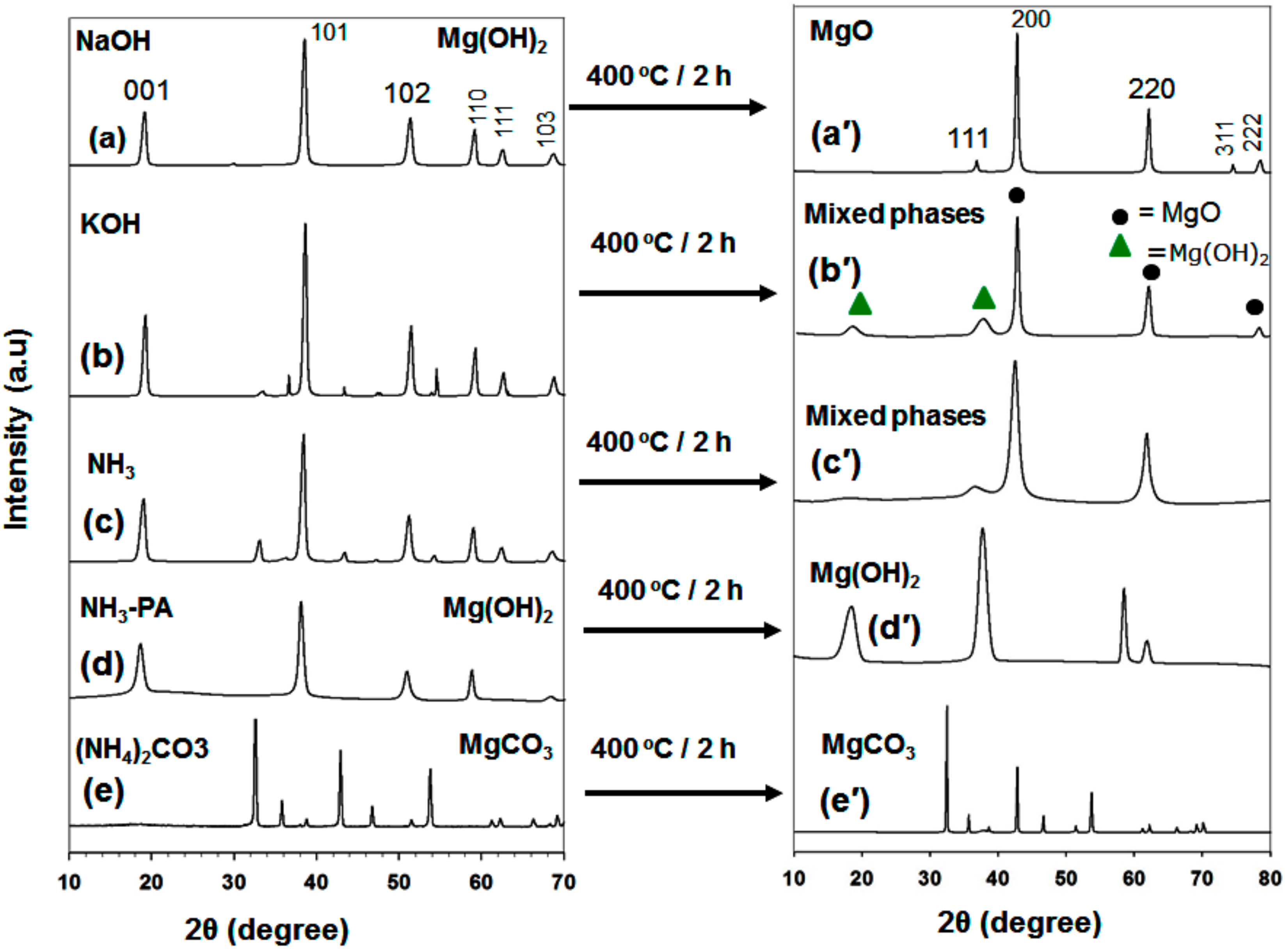
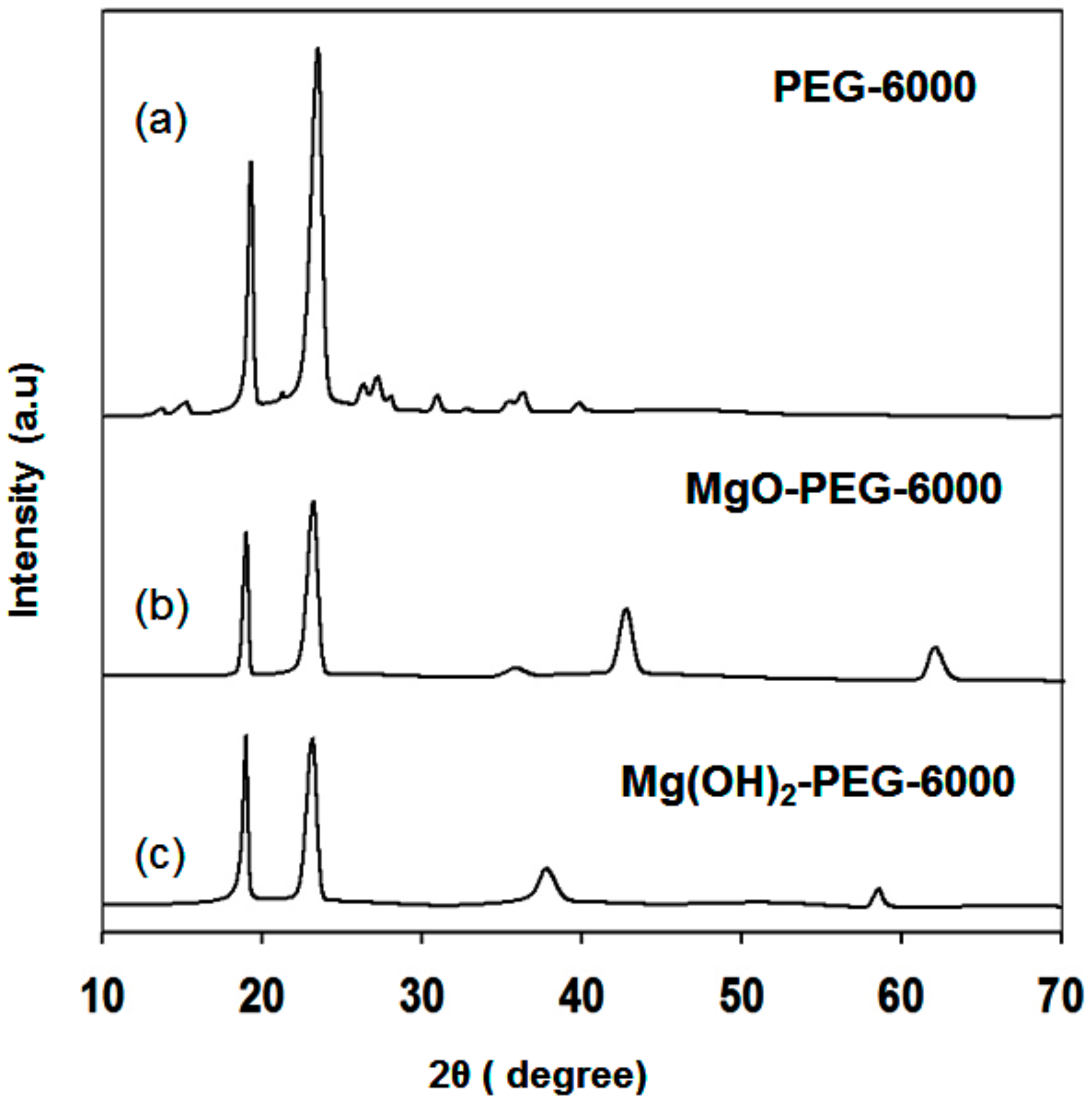
3.1.1. XRD Analysis
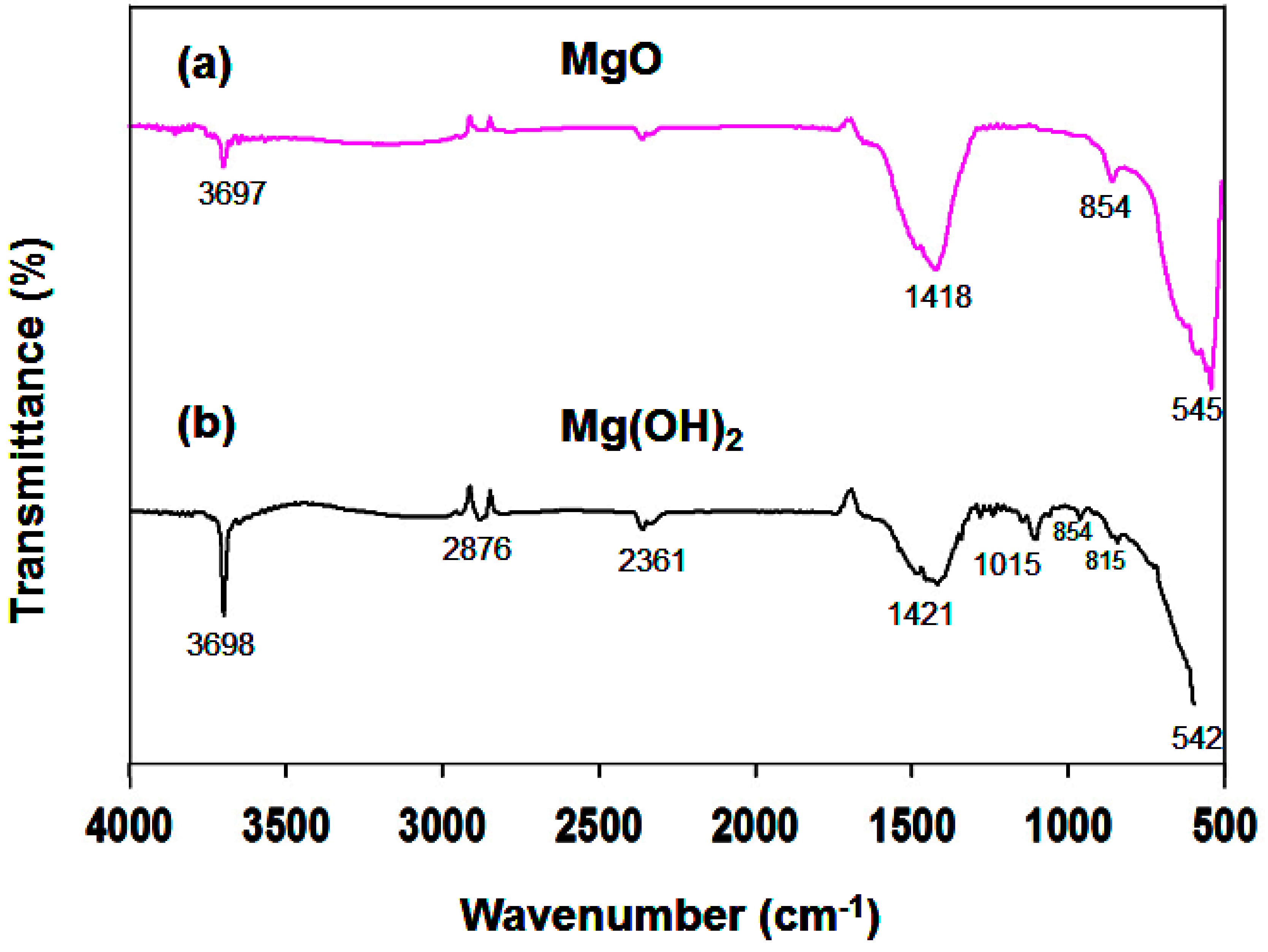
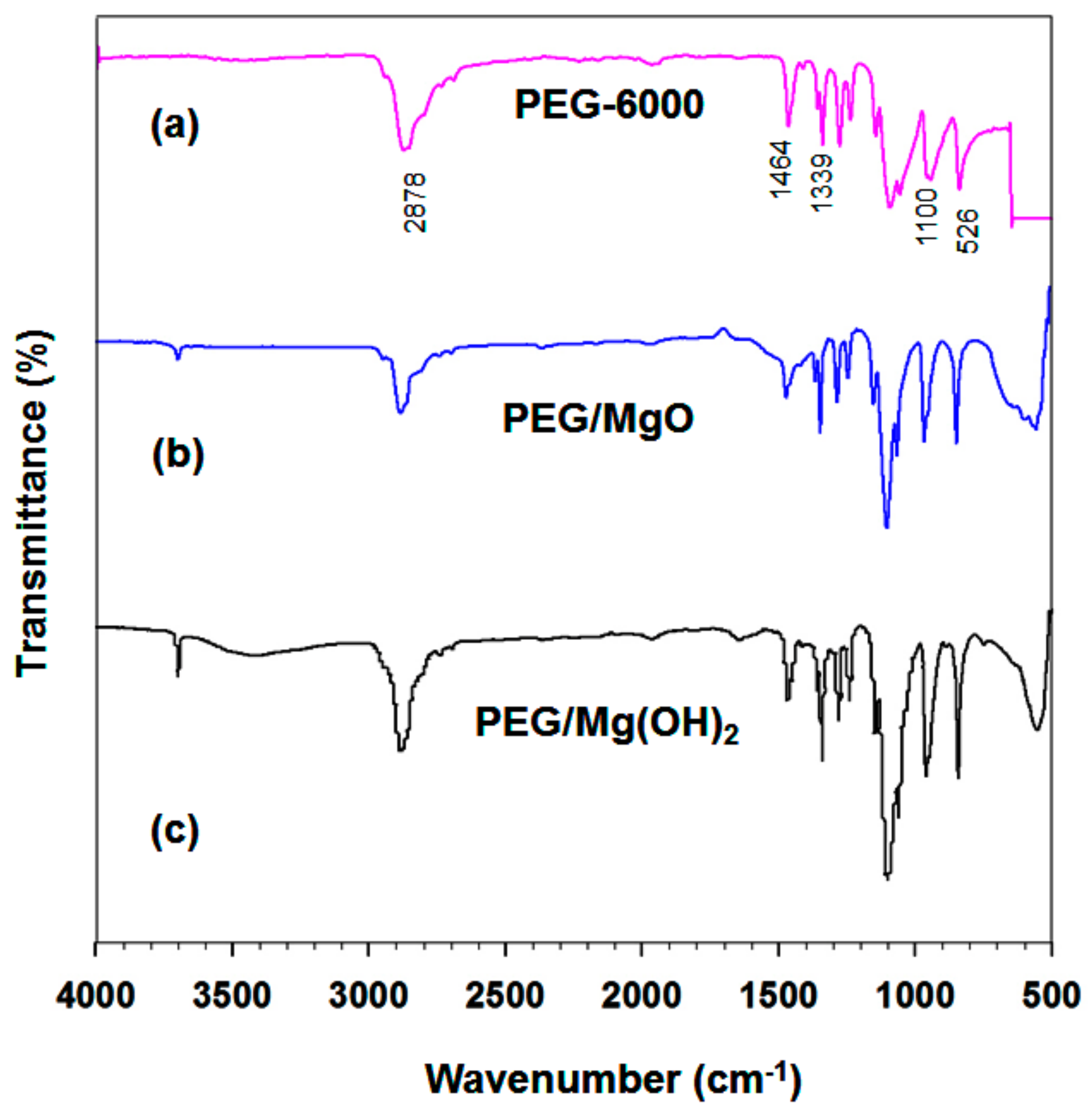
3.1.2. FTIR Analysis
3.1.3. Microstructures
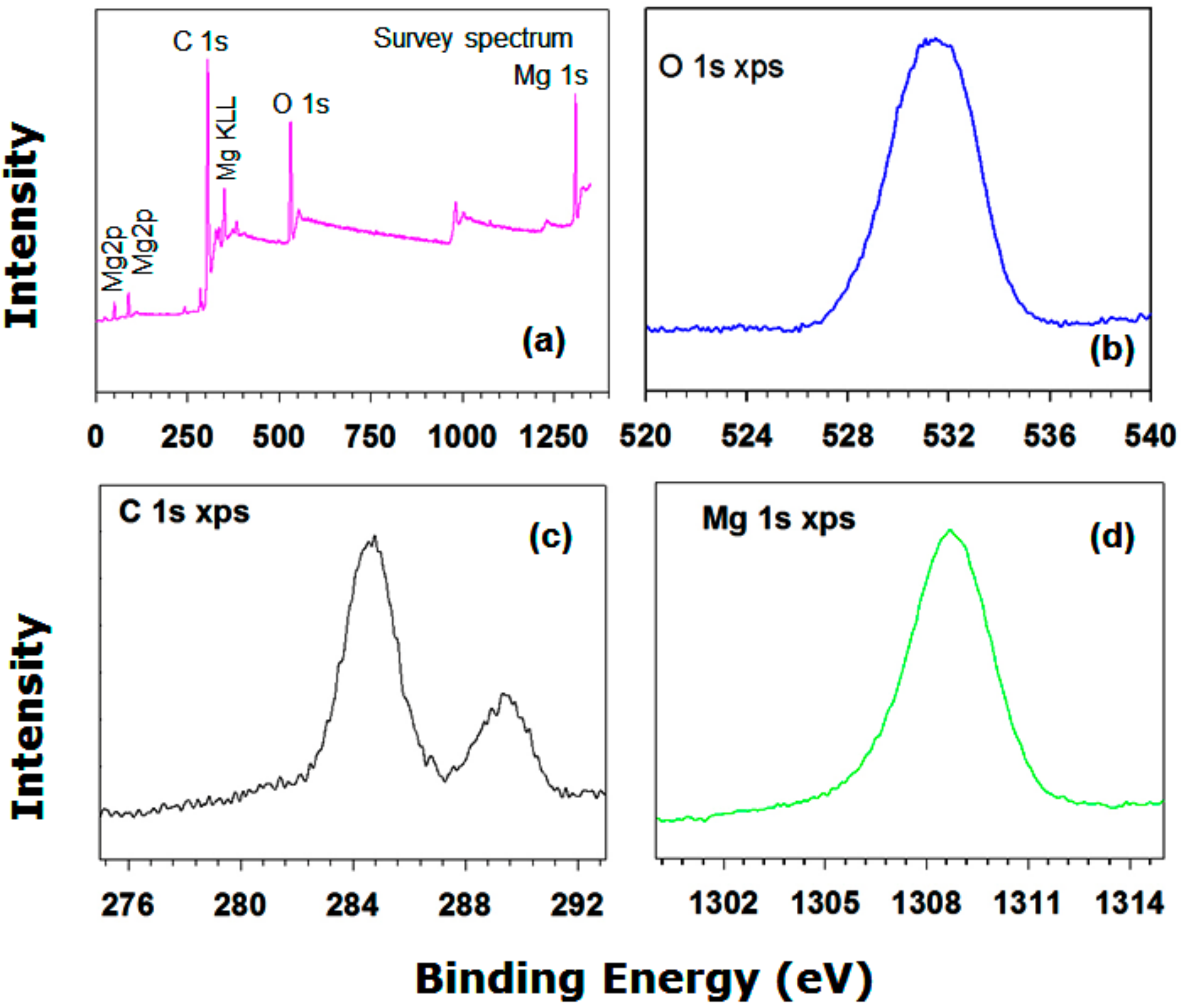
3.1.4. XPS Analysis
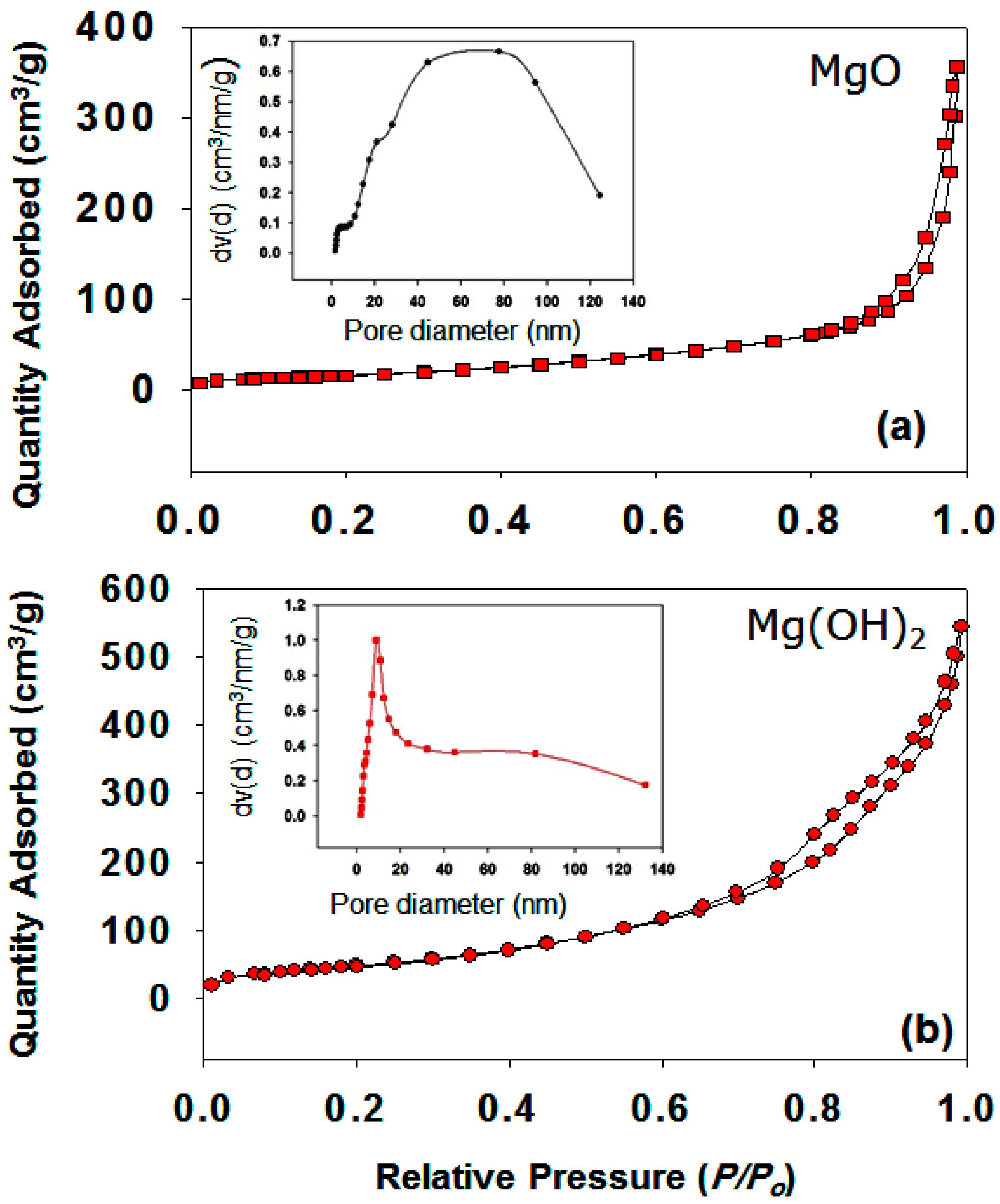
3.1.5. Effect of Pore Size Distribution and Pore Volume
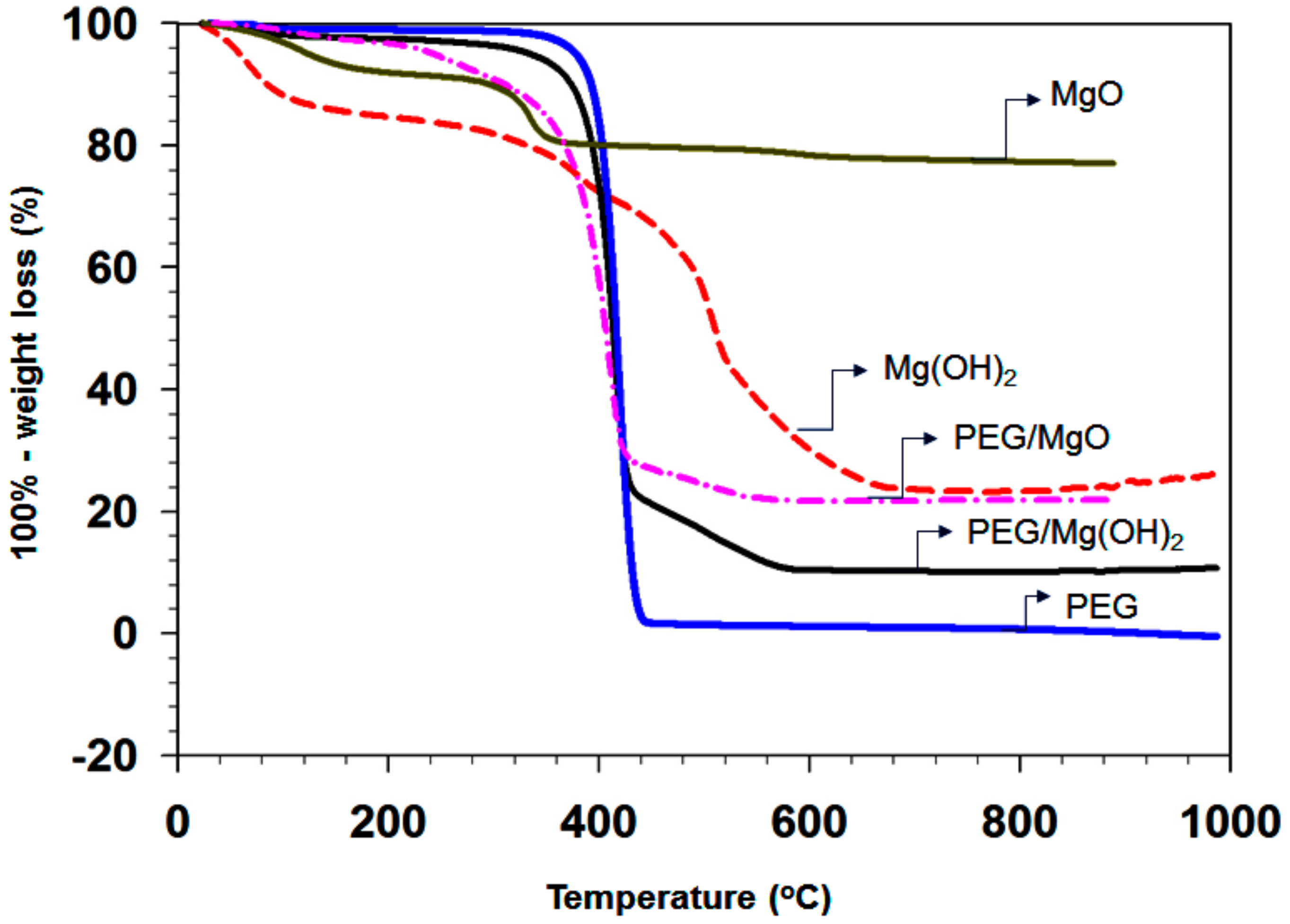
3.1.6. Thermal Stability of the Composites
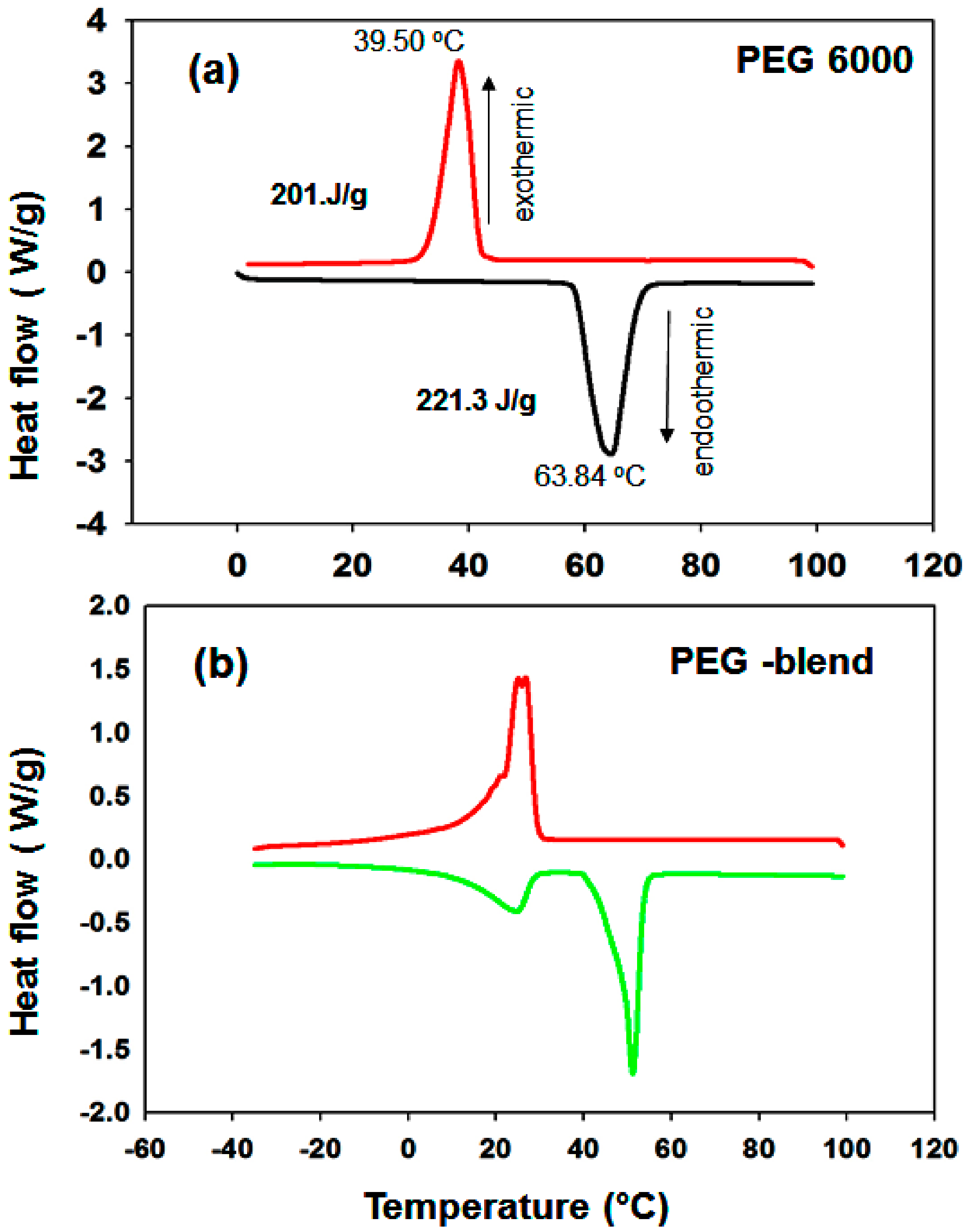
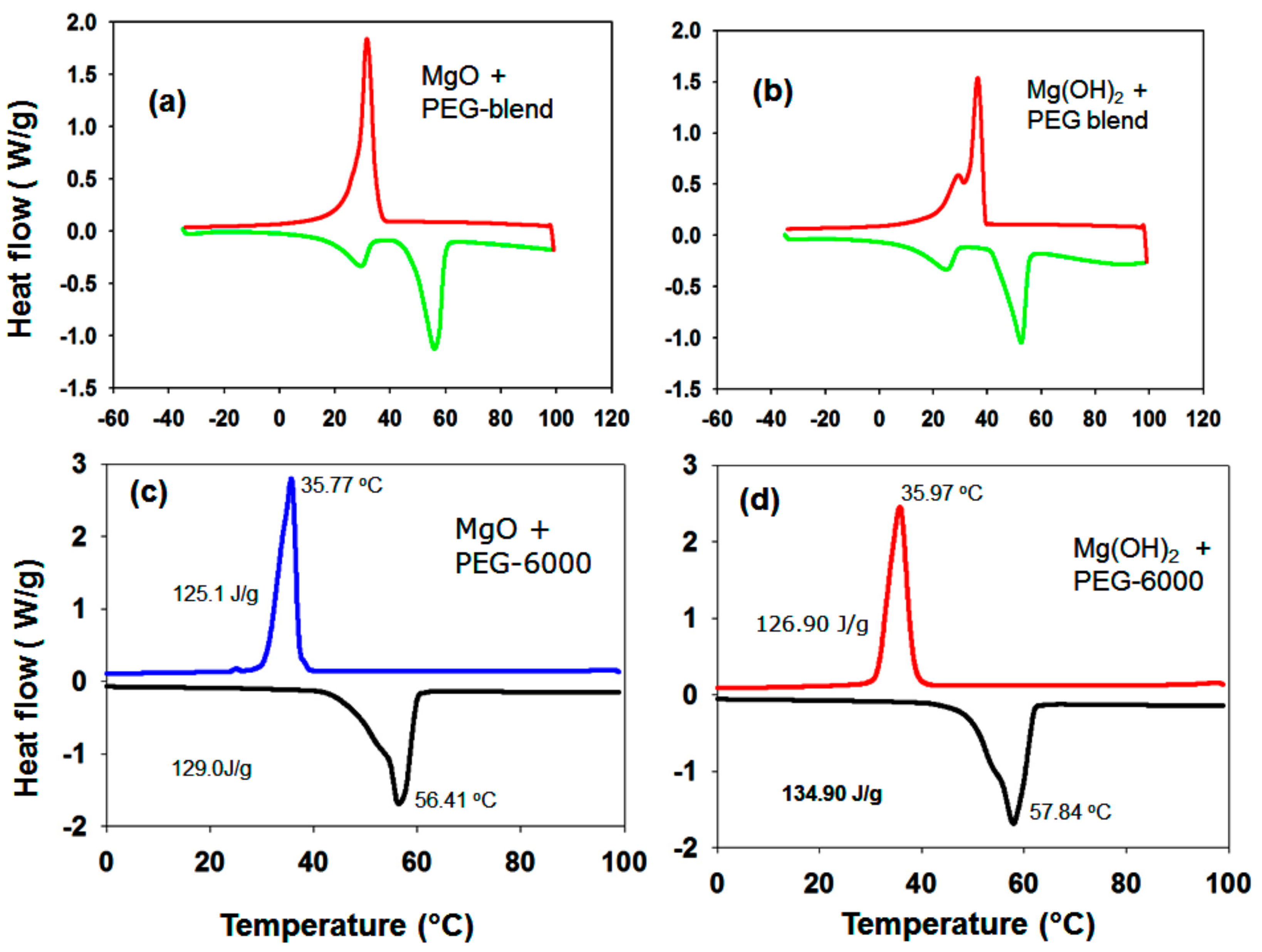
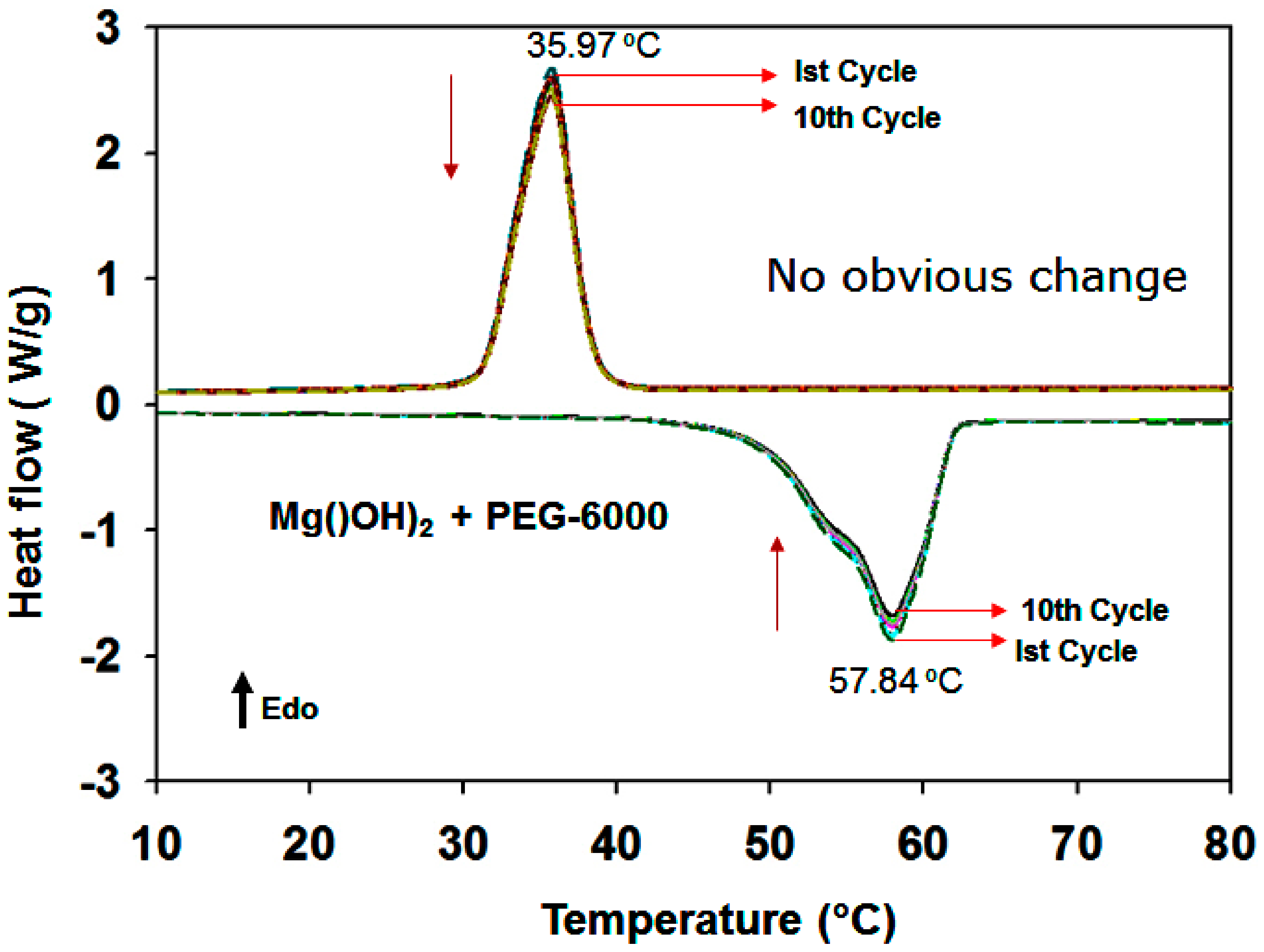
3.1.7. DSC Results of the PEG/MgO and PEG/Mg(OH)2 Composites
3.1.8. Thermal Conductivity
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sharma, A.; Tyagi, V.V.; Chen, C.R.; Buddhi, D. Review on thermal energy storage with phase change materials and applications. Renew. Sustain. Energy Rev. 2009, 13, 318–345. [Google Scholar] [CrossRef]
- Mohamed, S.A.; Al-Sulaiman, F.A.; Ibrahim, N.I.; Zahir, M.H.; Al-Ahmed, A.; Saidur, R.; Yılbaş, B.S.; Sahin, A.Z. A review on current status and challenges of inorganic phase change materials for thermal energy storage systems. Renew. Sustain. Energy Rev. 2016, 70, 1078–1089. [Google Scholar] [CrossRef]
- Kenisarin, M.; Mahkamov, K. Solar energy storage using phase change materials. Renew. Sustain. Energy Rev. 2007, 11, 1913–1965. [Google Scholar] [CrossRef]
- Zahir, M.H.; Mohamed, S.A.; Saidur, R.; Al-Sulaiman, F.A. Supercooling of Phase-Change materials and the techniques used to mitigate the phenomenon. Appl. Energy 2019, 240, 793–817. [Google Scholar] [CrossRef]
- Zhang, L.P.; Zhang, F.; Wang, M.; Kang, R.; Li, Y.; Mou, Y. Huang, Phase change materials based on polyethylene glycol supported by graphene-based mesoporous silica sheets. Appl. Therm. Eng. 2016, 101, 217–223. [Google Scholar] [CrossRef]
- Chen, C.; Liu, W.; Wang, Z.; Peng, K.; Xie, Q. Novel form stable phase change materials based on the composites of polyethylene glycol/polymeric solid-solid phase change material. Sol. Energy Mater. Sol. Cell. 2015, 134, 80–88. [Google Scholar] [CrossRef]
- Sharma, R.; Ganesan, P.; Tyagi, V.; Mahlia, T.M.I. Accelerated thermal cycle and chemical stability testing of polyethylene glycol (PEG) 6000 for solar thermal energy storage. Sol. Energy Mater. Sol. Cell. 2016, 147, 235–239. [Google Scholar] [CrossRef]
- Min, X.; Fang, M.; Huang, Z.; Huang, Y.; Wen, R.; Qian, T.; Wu, X. Enhanced thermal properties of novel shape-stabilized PEG composite phase change materials with radial mesoporous silica sphere for thermal energy storage. Sci. Rep. 2015, 5, 12964. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.W.; Li, Z.J. Paraffin/diatomite/multi-wall carbon nanotubes composite phase change material tailor-made for thermal energy storage cement-based composites. Energy 2014, 72, 371–380. [Google Scholar] [CrossRef]
- Tang, B.T.; Wu, C.; Qiu, M.G.; Zhang, X.W.; Zhang, S.F. PEG/SiO2-Al2O3 hybrid form stable phase change materials with enhanced thermal conductivity. Mater. Chem. Phys. 2014, 144, 162–167. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, S.Y.; Wang, J.P.; Yang, R. Preparation, stability and mechanical property of shape-stabilized phase change materials. Energy Build. 2014, 77, 11–16. [Google Scholar] [CrossRef]
- Karaman, S.; Karaipekli, A.; Sari, A.; Biçer, A. Polyethylene glycol (PEG)/diatomite composite as a novel form-stable phase change material for thermal energy storage. Sol. Energy Mater. Sol. Cell. 2011, 95, 1647–1653. [Google Scholar] [CrossRef]
- Sari, A.; Al-Sulaiman, F.A.; Zahir, M.H.; Al-Ahmed, A. Silica fume/capric acid-palmitic acid composite phase change material doped with CNTs for thermal energy storage. Sol. Energy Mater. Sol. Cell. 2017, 179, 361–363. [Google Scholar] [CrossRef]
- Li, W.; Zhang, R.; Jiang, N.; Tang, X.F. Composite macro capsule of phase change materials/expanded graphite for thermal energy storage. Energy 2013, 57, 607–614. [Google Scholar] [CrossRef]
- Guiyin, F.; Fang, T.; Lei, C. Preparation, thermal properties and applications of shape-stabilized thermal energy storage materials. Renew. Sustain. Energy Rev. 2014, 40, 237–259. [Google Scholar]
- Amin, M.; Putra, N.; Kosasih, E.A.; Prawiro, E.; Luanto, R.A.; Mahlia, T.M.I. Thermal properties of beeswax/graphene phase change material as energy storage for building applications. Appl. Therm. Eng. 2017, 112, 273–280. [Google Scholar] [CrossRef]
- Qian, T.T.; Li, J.; Ma, H.; Yang, J. The preparation of a green shape-stabilized composite phase change material of polyethylene glycol/SiO2 with enhanced thermal performance based on oil shale ash via temperature-assisted sol–gel method. Sol. Energy Mater. Sol. Cell. 2015, 132, 29–39. [Google Scholar] [CrossRef]
- Tang, B.T.; Cui, J.S.; Wang, Y.M.; Jia, C.; Zhang, S.F. Facile synthesis and performances of PEG/SiO2 composite form-stable phase change materials. Sol. Energy 2013, 97, 484–492. [Google Scholar] [CrossRef]
- Py, X.; Olives, R.; Mauran, S. Paraffin/porous-graphite-matrix composite as a high and constant power thermal storage material. Int. J. Heat Mass Transf. 2001, 44, 2727–2737. [Google Scholar] [CrossRef]
- Feng, L.; Zhao, W.; Zheng, J.; Frisco, S.; Song, P.; Li, X. The shape-stabilized phase change materials composed of polyethylene glycol and various mesoporous matrices (AC,SBA-15 and MCM-41). Sol. Energy Mater. Sol. Cell. 2011, 95, 3550–3556. [Google Scholar] [CrossRef]
- Shudong, Z.; Min, Z.; Xia, L.; Changzheng, W.; Yongfu, S.; Yi, X. Macroscaled mesoporous calcium carbonate tetragonal prisms: Top-down solid-phase fabrication and applications of phase-change material support matrices. CrysEngComm 2010, 12, 3571–3578. [Google Scholar]
- Tingting, Q.; Jinhong, L.; Xin, M.; Yong, D.; Weimin, G.; Hongwen, M. Polyethylene glycol/mesoporous calcium silicate shape-stabilized composite phase change material: Preparation, characterization, and adjustable thermal property. Energy 2015, 82, 333–340. [Google Scholar]
- Hao, Y.; Shao, X.; Liu, T.; Li, B.; Nie, S. Porous MgO material with ultrahigh surface area as the matrix for phase change composite. Thermochim. Acta 2015, 604, 45–51. [Google Scholar] [CrossRef]
- Tingting, Q.; Jinhong, L.; Xin, M.; Weimin, G.; Yong, D.; Lei, N. Enhanced thermal conductivity of PEG/diatomite shape-stabilized phase change materials with Ag nanoparticles for thermal energy storage. J. Mater. Chem. A 2015, 3, 8526–8536. [Google Scholar]
- Chaoen, L.; Hang, Y.; Yuan, S.; Mei, Z. Synthesis and characterization of PEG/ZSM-5 composite phase change, materials for latent heat storage. Renew. Energy 2018, 121, 45–52. [Google Scholar]
- Yuang, Z.; Jiasheng, W.; Jinjing, Q.; Xin, J.; Malik, M.U.; Rongwen, L.; Shufen, Z.; Bingtao, T. Ag-graphene/PEG composite phase change materials for enhancing solarthermal energy conversion and storage capacity. Appl. Energy 2019, 237, 83–90. [Google Scholar]
- Yang, H.Z.; Feng, L.L.; Wang, C.Y.; Zhao, W.; Li, X.G. Confinement effect of SiO2 framework on phase change of PEG in shape-stabilized PEG/SiO2 composites. Eur. Polym. J. 2012, 48, 803–810. [Google Scholar] [CrossRef]
- Weilong, W.; Xiaoxi, Y.; Yutang, F.; Jing, D. Preparation and performance of form-stable polyethylene glycol/silicon dioxide composites as solid–liquid phase change materials. Appl. Energy 2009, 86, 170–174. [Google Scholar]
- Yuping, W.; Tao, W. The dependence of phase change enthalpy on the pore structure and interfacial groups in hydrated salts/silica composites via sol–gel. J. Colloid Interface Sci. 2015, 448, 100–105. [Google Scholar]
- Slifka, A.J.; Filla, B.J.; Phelps, J.M. Thermal Conductivity of Magnesium Oxide from Absolute, Steady-State Measurements. J. Res. Natl. Inst. Stand. Technol. 1998, 103, 357–363. [Google Scholar] [CrossRef]
- Yang, L.; May, P.W.; Yin, L.; Smith, J.A.; Rosser, K.N. Growth of diamond nanocrystals by pulse laser ablation of graphite in liquid. Diamond Relat. Mater. 2007, 16, 725–729. [Google Scholar] [CrossRef]
- Tran, X.P.; Bret, H.; Martello, D.V.; Yee, S. Synthesis of Mg(OH)2, MgO, and Mg nanoparticles using laser ablation of magnesium in water and solvents. Opt. Laser. Eng. 2008, 46, 829–834. [Google Scholar]
- Aramendia, M.; Borau, V.; Jimenez, C. Synthesis and characterization of various MgO and related systems. J. Mater. Chem. 1996, 6, 1943–1949. [Google Scholar] [CrossRef]
- Cui, H.; Wu, X.; Chen, Y.; Boughton, R.I. Synthesis and characterization of mesoporous MgO by template-free hydrothermal method. Mater. Res. Bull. 2014, 50, 307–311. [Google Scholar] [CrossRef]
- Kamimoto, T.M.; Abe, Y.; Sakamoto, R.; Kanari, K.; Ozawa, T. Investigation of latent heat-thermal energy storage materials. iv. Thermos analytical evaluation of binary eutectic mixtures of NaOH with LiOH or KOH. Thermochimica Acta 1987, 121, 193–202. [Google Scholar]
- Balducci, G.; Diaz, L.B.; Gregory, D.H. Recent progress in the synthesis of nanostructured magnesium hydroxide. CrystEngComm 2017, 19, 6067–6084. [Google Scholar] [CrossRef] [Green Version]
- Sulaiman, K.O.; Aziz, A.; Zahir, M.H.; Shaikh, M.N.; Al-Betar, A.R.; Oyama, M. Hydrothermal synthesis of tin-doped indium oxide nanoparticles using pamoic acid as an organic additive and their photoluminescence properties. J. Mater. Sci. Mater. Electron. 2017, 28, 3226–3233. [Google Scholar]
- Yi, D.; Guangtao, Z.; Hao, W.; Bin, H.; Liangbin, W.; Yitai, Q. Nanoscale Magnesium Hydroxide and Magnesium Oxide Powders: Control over Size, Shape, and Structure via Hydrothermal Synthesis. Chem. Mater. 2001, 13, 435–440. [Google Scholar]
- Wang, C.; Feng, L.; Li, W.; Zheng, J.; Tian, W.; Li, X. Shape-stabilized phase change materials based on polyethylene glycol/porous carbon composite: The influence of the pore structure of the carbon materials. Sol. Energy Mater. Sol. Cell. 2012, 105, 21–26. [Google Scholar] [CrossRef]
- Agnieszka, A.P.; Łukasz, K.; Teofil, J. Recent development in the synthesis, modification and application of Mg(OH)2 and MgO: A review. Powder Technol. 2017, 319, 373–407. [Google Scholar]
- Agnieszka, P.; Iwona, L.; Marcin, W.; Dominik, P.; Teofil, J. Synthesis of Mg(OH)2 from magnesium salts and NH4OH by direct functionalization with poly(ethylene glycols). Physicochem. Probl. Miner. Process. 2012, 48, 631–643. [Google Scholar]
- Hu, N.; Wu, Z.P.; Wang, G.W.; Wen, P.; Qian, W. Preparation of Ultrafine Magnesium Hydroxide with Two Kinds of Dispersing Agent. In Proceedings of the 1st International Conference on Mechanical Engineering and Material Science (MEMS), Shanghai, China, 28–30 December 2012; Series: Advances in Intelligent Systems Research, Conference Proceeding. AISR: Seoul, Korea, 2012; Volume 27. [Google Scholar] [CrossRef] [Green Version]
- Hai, C.; Li, S.; Zhou, Y.; Zeng, J.; Ren, X.; Li, X. Roles of ethylene glycol solvent and polymers in preparing uniformly distributed MgO nanoparticles. J. Asian Ceram. Soc. 2017, 5, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Niazi, S.K. Handbook of Pharmaceutical Manufacturing Formulations: Semisolid Products; CRC Press: New York, NY, USA, 2009; Available online: https://kampoeng2013.files.wordpress.com/2016/10/handbook-of-pharmaceutical-manufacturing-formulations-semisolid-preparation.pdf (accessed on 12 December 2019).
- Wang, W.; Yang, X.; Fang, Y.; Ding, J.; Yan, J. Enhanced thermal conductivity and thermal performance of form-stable composite phase change materials by using ß-Aluminum nitride. Appl. Energy 2009, 86, 1196–1200. [Google Scholar] [CrossRef]
- Zahir, M.H.; Irshad, K.; Aziz, M.A.; Md Shafiullah, M.; Rahman, M.M.; Hossain, M.M. Shape-Stabilized Phase Change Material for Solar Thermal Energy Storage: CaO Containing MgCO3 Mixed with Polyethylene Glycol. Energy Fuels 2019, 33, 12041–12051. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Results | Samples | |
|---|---|---|
| MgO | Mg(OH)2 | |
| Specific surface area (m2g−1) | 63.0024 | 184.9750 |
| Pore volume (ccg−1) | 0.5519 | 0.8421 |
| Mean pore diameter (nm) | 20.2913 | 10.5147 |
| Sample | Tf (°C) | Tm (°C) | ΔHf (J/g) | ΔHm (J/g) | ΔTs | R (%) | E (%) | φ (%) |
|---|---|---|---|---|---|---|---|---|
| * PEG (6000) | 39.5 | 63.84 | 201.0 | 221.3 | 24.34 | - | ||
| ** PEG-6000/MgO | 35.77 | 56.41 | 110.4 | 129.0 | 20.64 | 58.29 | 56.69 | 97.25 |
| *** PEG-6000/Mg(OH)2 | 37.97 | 57.84 | 118.5 | 134.90 | 19.89 | 60.96 | 60.00 | 98.43 |
| 1 PEG1000/MgO | 18.30 | 34.40 | - | 61.62 | 16.10 | 64.6 | ||
| 2 PEG-10,000/SiO2 | - | 61.61 | - | 162.9 | - | - | ||
| 3 PEG-1000/SiO2-ß-AIN | 45.13 | 60.41 | 161.4 | 132.9 | 15.28 | - | ||
| 4 PEG/CaO4Si-6000 | 44.19 | 57.59 | 99.53 | 113.60 | 13.40 | - |
| Sample | Conductivity (Wm−1K−1) |
|---|---|
| PEG-6000 | 0.2124 |
| PEG-6000/MgO | 0.7423 |
| PEG-6000/Mg(OH)2 | 0.3334 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahir, M.H.; Rahman, M.M.; Irshad, K.; Rahman, M.M. Shape-Stabilized Phase Change Materials for Solar Energy Storage: MgO and Mg(OH)2 Mixed with Polyethylene Glycol. Nanomaterials 2019, 9, 1773. https://doi.org/10.3390/nano9121773
Zahir MH, Rahman MM, Irshad K, Rahman MM. Shape-Stabilized Phase Change Materials for Solar Energy Storage: MgO and Mg(OH)2 Mixed with Polyethylene Glycol. Nanomaterials. 2019; 9(12):1773. https://doi.org/10.3390/nano9121773
Chicago/Turabian StyleZahir, Md. Hasan, Mohammad Mizanur Rahman, Kashif Irshad, and Mohammad Mominur Rahman. 2019. "Shape-Stabilized Phase Change Materials for Solar Energy Storage: MgO and Mg(OH)2 Mixed with Polyethylene Glycol" Nanomaterials 9, no. 12: 1773. https://doi.org/10.3390/nano9121773