

Degradation of Phenol via an Advanced Oxidation Process (AOP) with Immobilized Commercial Titanium Dioxide (TiO2) Photocatalysts
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Photocatalysts
2.2. Immobilization of the TiO2 Photocatalyst Particles
2.3. Degradation of Phenol
2.4. Data Evaluation
2.5. Analytic Methods
2.5.1. HPLC
2.5.2. TOC
2.5.3. SEM
2.5.4. SEM-EDX Mapping
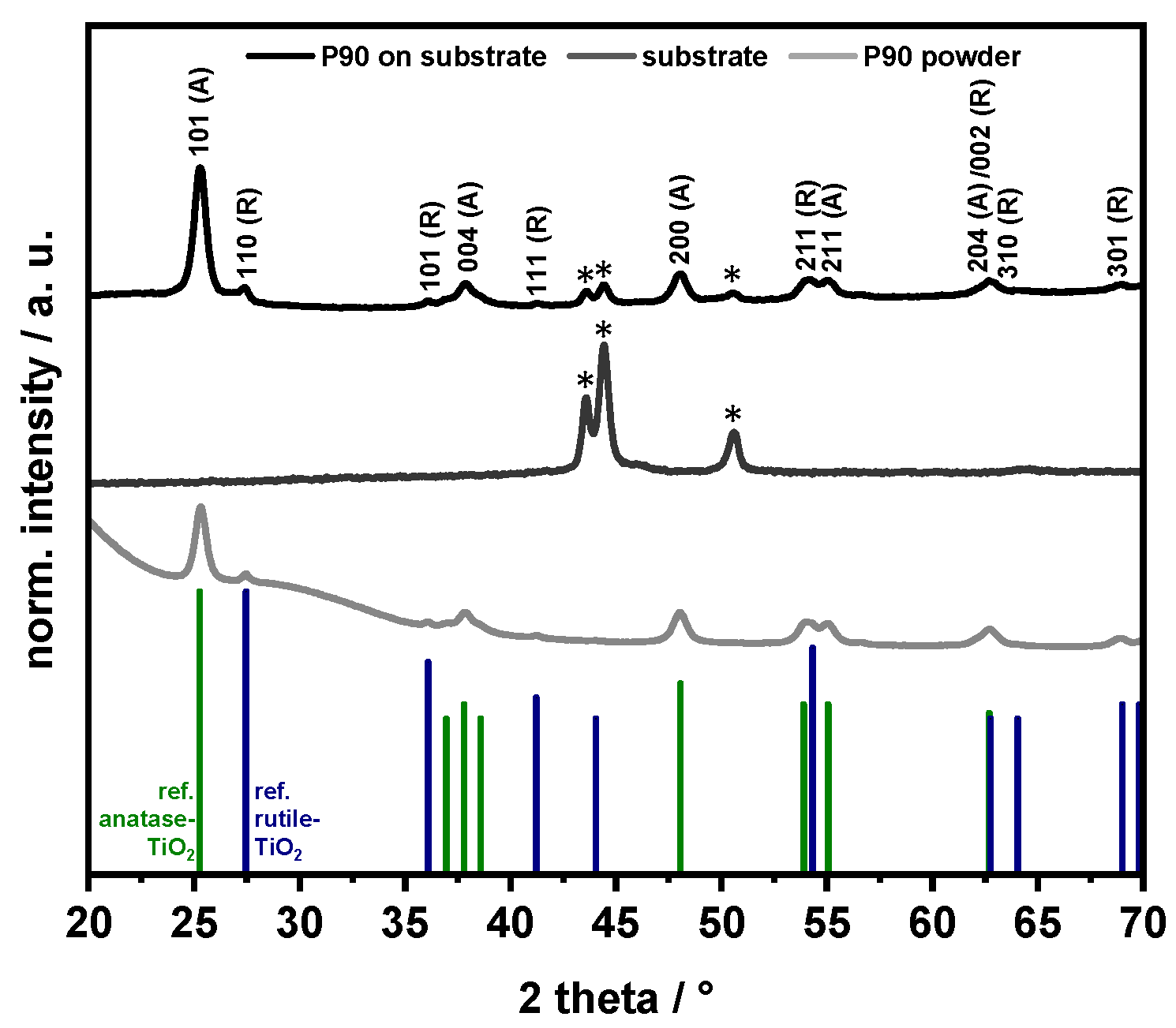
2.5.5. XRD
3. Results and Discussion
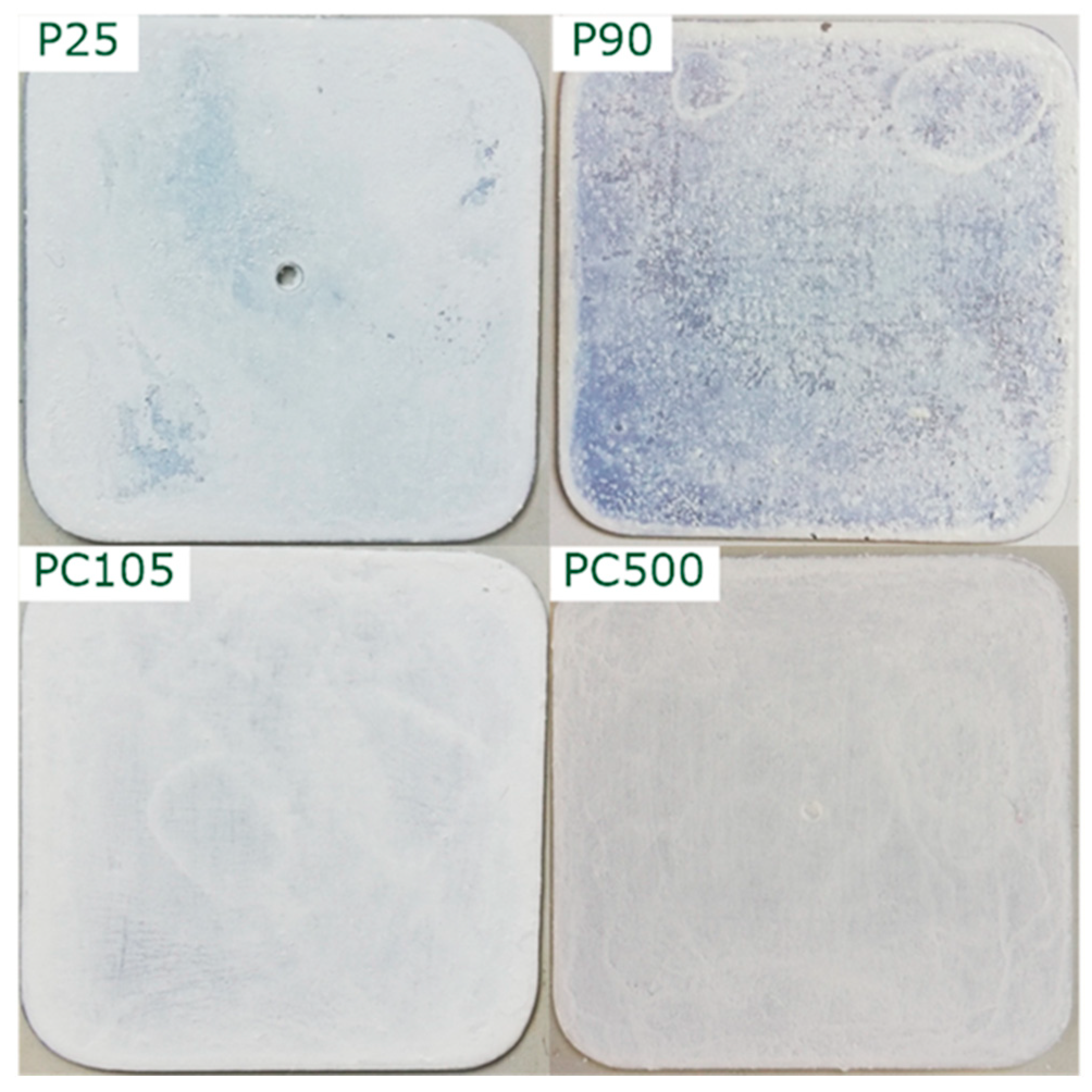
3.1. Characterization of the TiO2 Films
3.2. Degradation of Phenol with Immobilized TiO2 Photocatalyst Particles
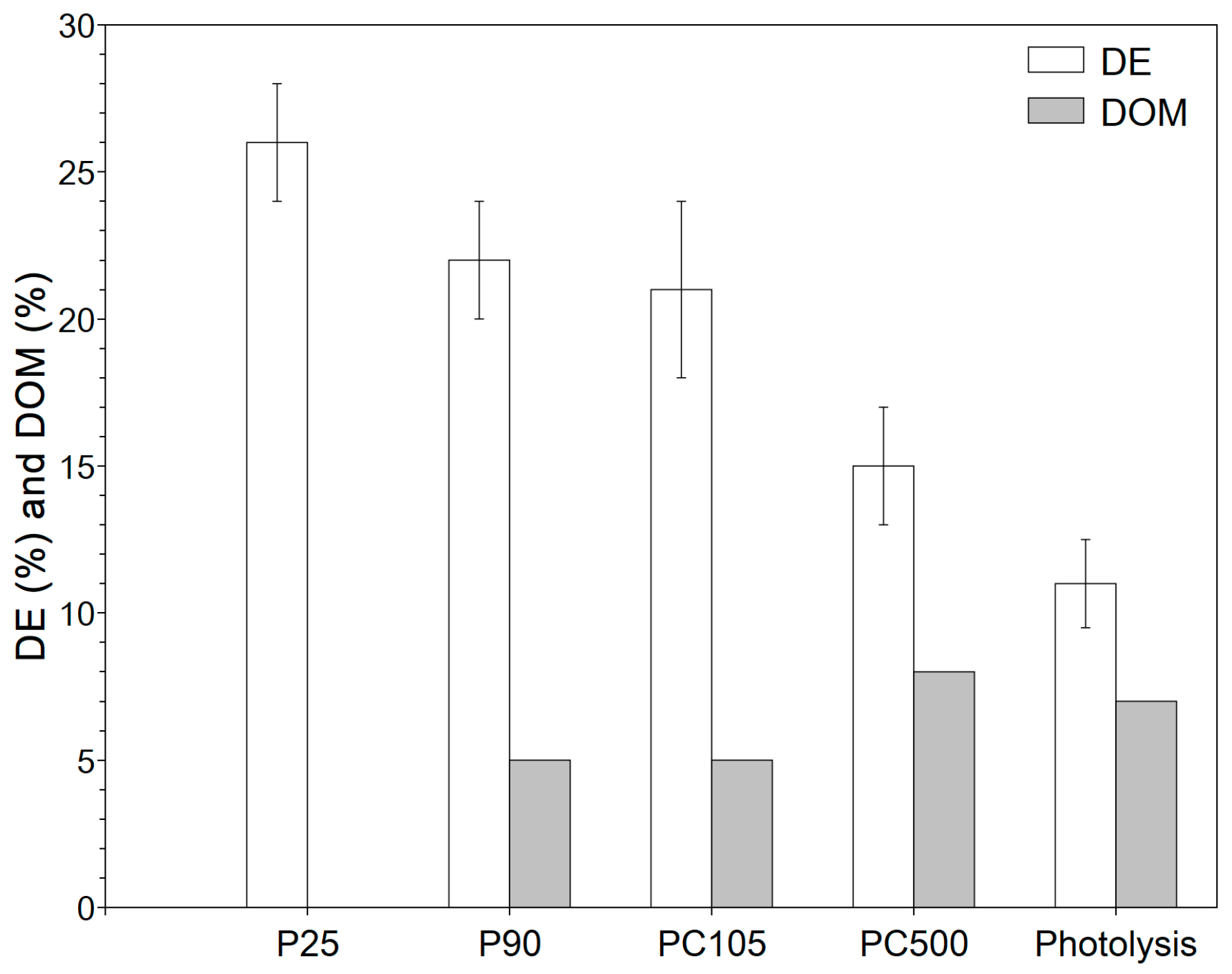
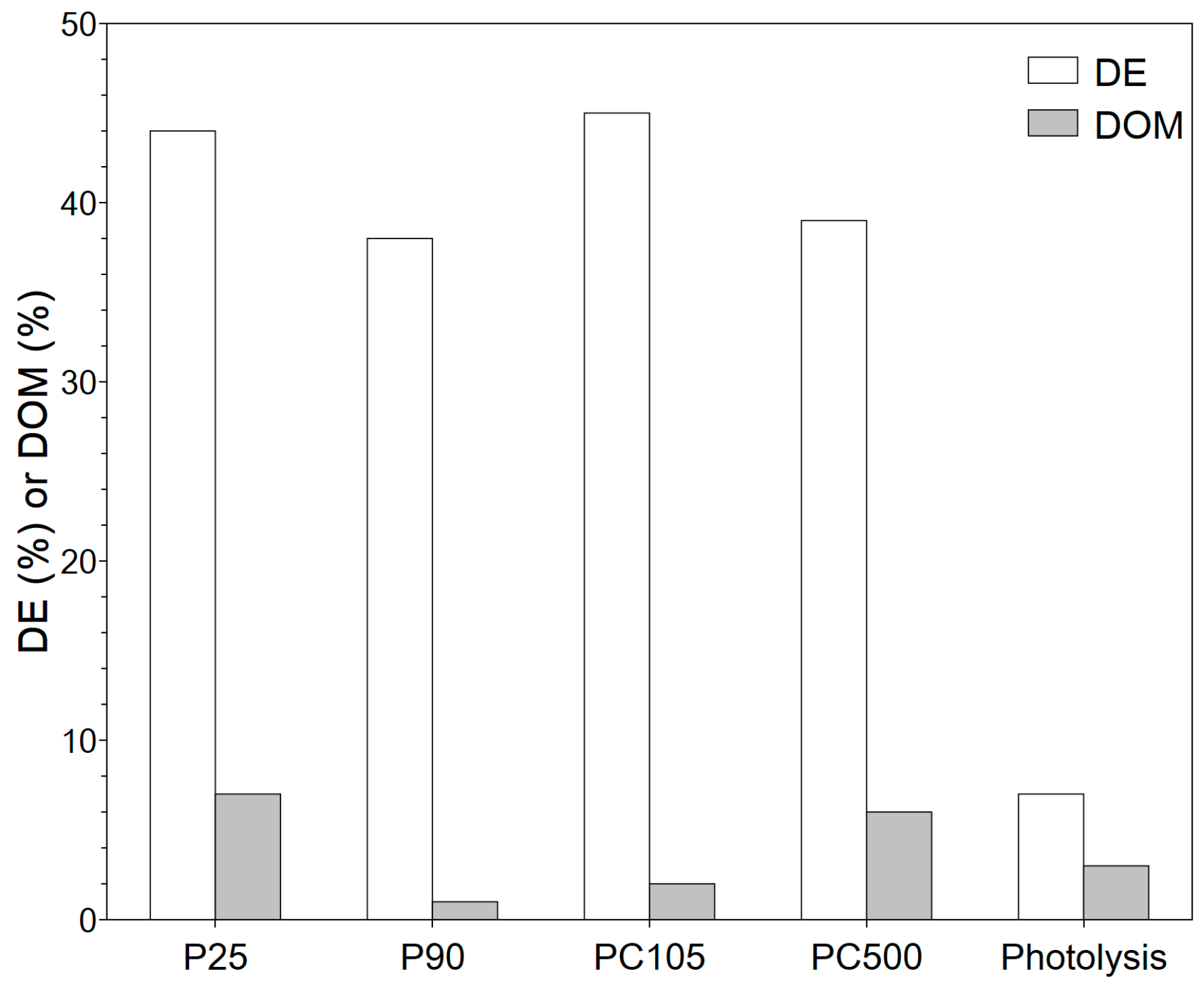
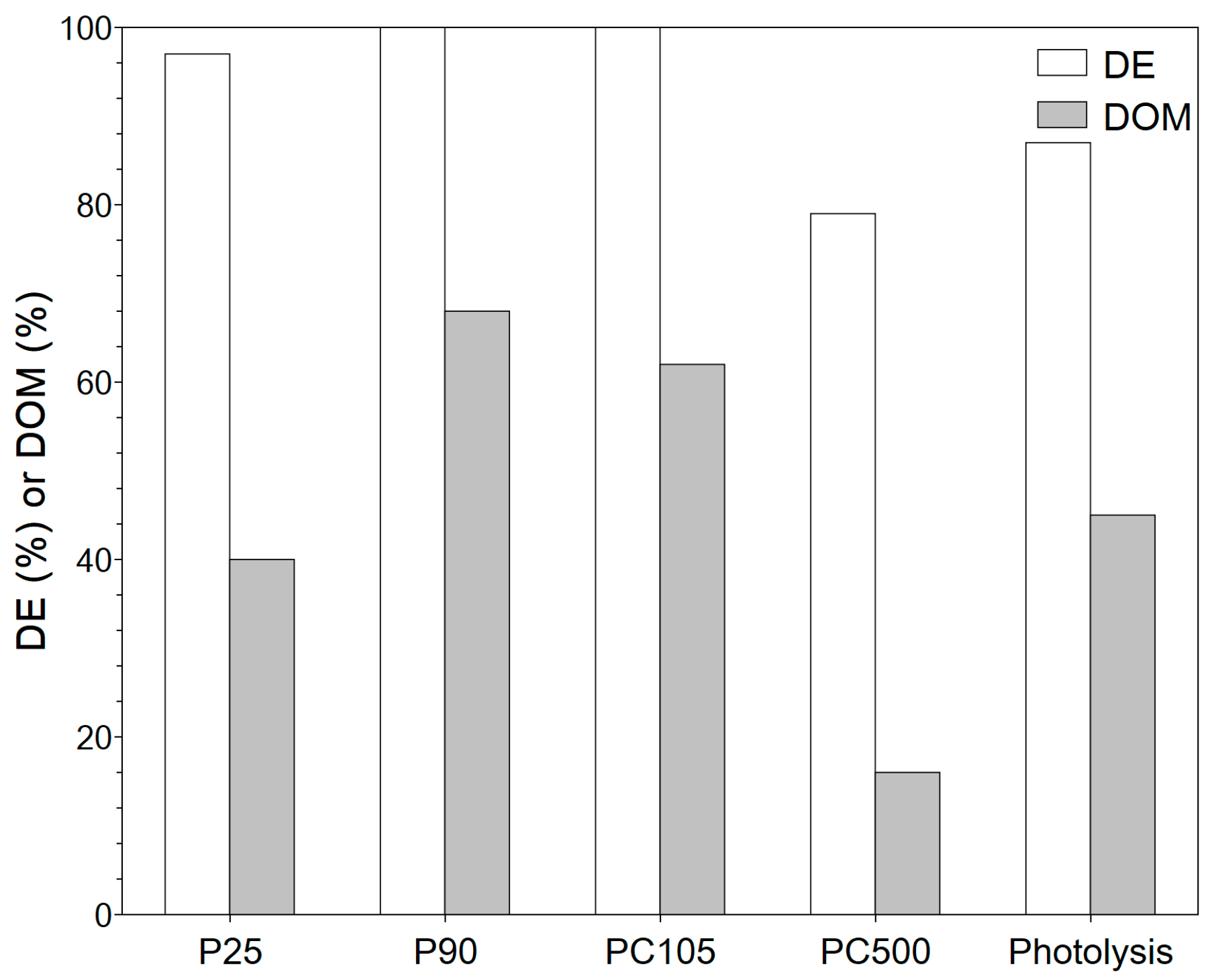
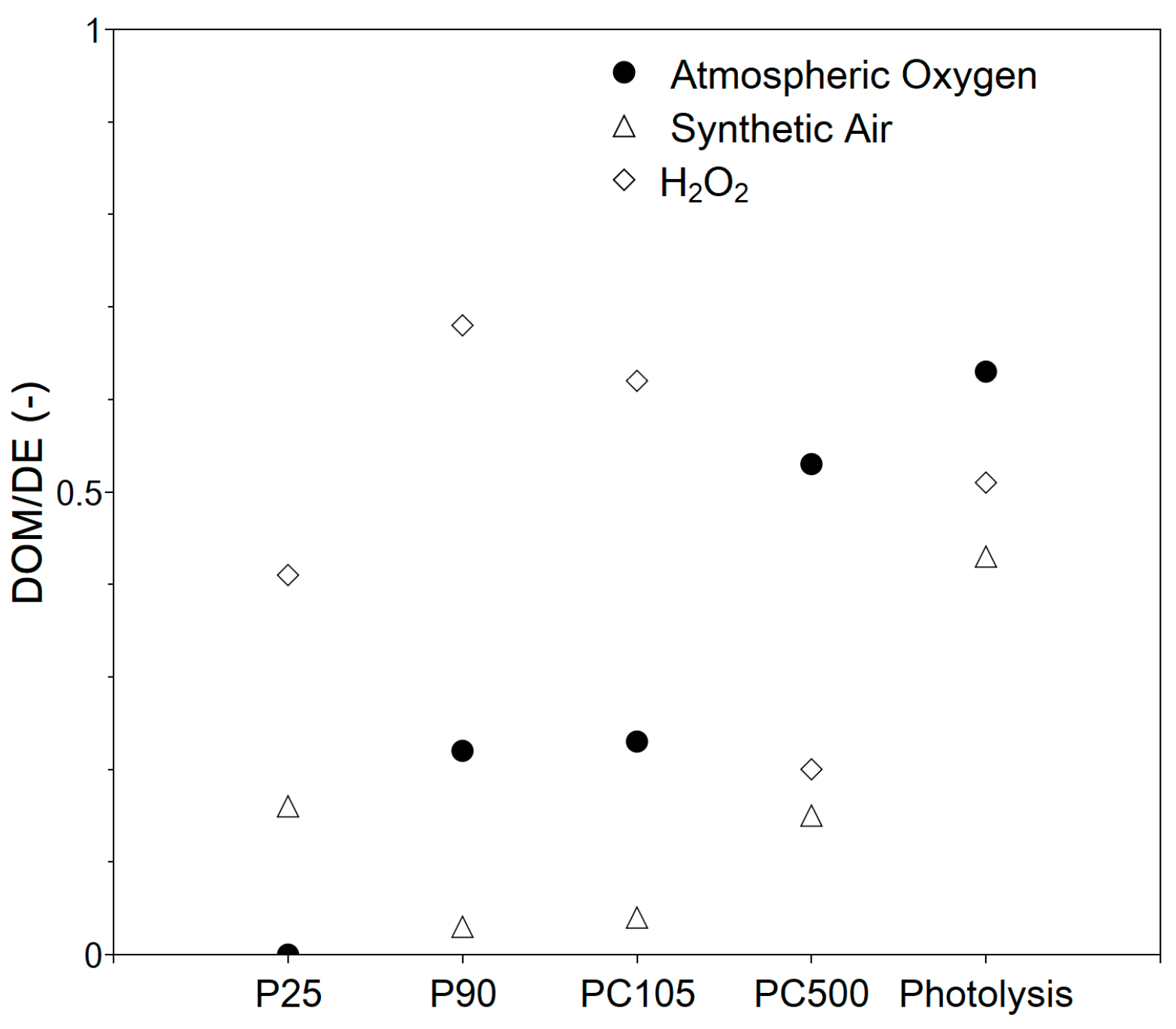
3.2.1. System Selection
3.2.2. Impact of Operating Conditions
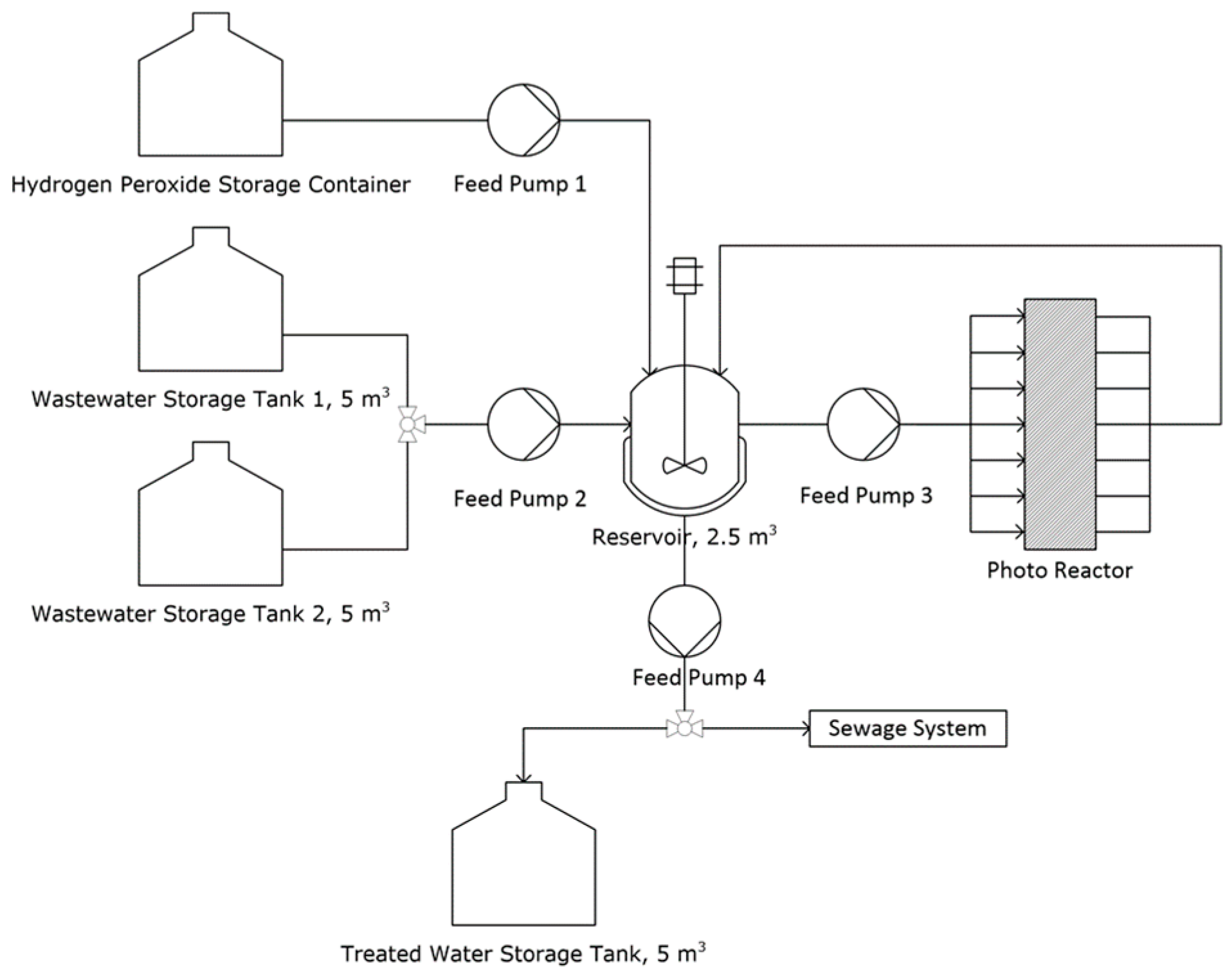
3.3. Plant Design and Cost Estimation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- El-Naas, M.H.; Al-Zuhair, S.; Alhaija, M.A. Removal of phenol from petroleum refinery wastewater through adsorption on date-pit activated carbon. Chem. Eng. J. 2010, 162, 997–1005. [Google Scholar] [CrossRef]
- Pradeep, N.V.; Anupama, S.; Navya, K.; Shalini, H.N.; Idris, M.; Hampannavar, U.S. Biological removal of phenol from wastewaters: A mini review. Appl. Water Sci. 2015, 5, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, S.; Kargari, A.; Sanaeepur, H.; Abbassian, K.; Najafi, A.; Mofarrah, E. Phenol removal from industrial wastewaters: A short review. Desalin. Water Treat. 2015, 53, 2215–2234. [Google Scholar] [CrossRef]
- Villegas, L.G.C.; Mashhadi, N.; Chen, M.; Mukherjee, D.; Taylor, K.E.; Biswas, N. A Short Review of Techniques for Phenol Removal from Wastewater. Curr. Pollut. Rep. 2016, 2, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, S. Reactive species in advanced oxidation processes: Formation, identification and reaction mechanism. Chem. Eng. J. 2020, 401, 126158. [Google Scholar] [CrossRef]
- Suzuki, H.; Araki, S.; Yamamoto, H. Evaluation of advanced oxidation processes (AOP) using O3, UV, and TiO2 for the degradation of phenol in water. J. Water Process Eng. 2015, 7, 54–60. [Google Scholar] [CrossRef]
- Miklos, D.B.; Remy, C.; Jekel, M.; Linden, K.G.; Drewes, J.E.; Hübner, U. Evaluation of advanced oxidation processes for water and wastewater treatment—A critical review. Water Res. 2018, 139, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhao, R. Advanced Oxidation Processes (AOPs) in Wastewater Treatment. Curr. Pollut. Rep. 2015, 1, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Berkani, M.; Smaali, A.; Kadmi, Y.; Almomani, F.; Vasseghian, Y.; Lakhdari, N.; Alyane, M. Photocatalytic degradation of Penicillin G in aqueous solutions: Kinetic, degradation pathway, and microbioassays assessment. J. Hazard. Mater. 2022, 421, 126719. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Gao, P.; Wang, L.; Zhang, Y.; Fu, J.; Huang, R.; Zhao, S.; Wang, G.; Wei, Y.; Zhou, S. Activation of peroxymonosulfate by MnO2 with oxygen vacancies: Degradation of organic compounds by electron transfer nonradical mechanism. J. Environ. Chem. Eng. 2022, 10, 107481. [Google Scholar] [CrossRef]
- Gehrke, I.; Geiser, A.; Somborn-Schulz, A. Innovations in nanotechnology for water treatment. Nanotechnol. Sci. Appl. 2015, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostami, M.; Badiei, A.; Ganjali, M.R.; Rahimi-Nasrabadi, M.; Naddafi, M.; Karimi-Maleh, H. Nano-architectural design of TiO2 for high performance photocatalytic degradation of organic pollutant: A review. Environ. Res. 2022, 212, 113347. [Google Scholar] [CrossRef] [PubMed]
- Eddy, D.R.; Ishmah, S.N.; Permana, M.D.; Firdaus, M.L.; Rahayu, I.; El-Badry, Y.A.; Hussein, E.E.; El-Bahy, Z.M. Photocatalytic Phenol Degradation by Silica-Modified Titanium Dioxide. Appl. Sci. 2021, 11, 9033. [Google Scholar] [CrossRef]
- Ahmed, S.; Rasul, M.G.; Martens, W.N.; Brown, R.; Hashib, M.A. Advances in Heterogeneous Photocatalytic Degradation of Phenols and Dyes in Wastewater: A Review. Water Air Soil Pollut. 2011, 215, 3–29. [Google Scholar] [CrossRef] [Green Version]
- Chiou, C.-H.; Wu, C.-Y.; Juang, R.-S. Influence of operating parameters on photocatalytic degradation of phenol in UV/TiO2 process. Chem. Eng. J. 2008, 139, 322–329. [Google Scholar] [CrossRef]
- Houas, A. Photocatalytic degradation pathway of methylene blue in water. Appl. Catal. B Environ. 2001, 31, 145–157. [Google Scholar] [CrossRef]
- Wang, Y. Solar photocatalytic degradation of eight commercial dyes in TiO2 suspension. Water Res. 2000, 34, 990–994. [Google Scholar] [CrossRef]
- Jallouli, N.; Pastrana-Martínez, L.M.; Ribeiro, A.R.; Moreira, N.F.F.; Faria, J.L.; Hentati, O.; Silva, A.M.T.; Ksibi, M. Heterogeneous photocatalytic degradation of ibuprofen in ultrapure water, municipal and pharmaceutical industry wastewaters using a TiO2/UV-LED system. Chem. Eng. J. 2018, 334, 976–984. [Google Scholar] [CrossRef]
- Yang, H.; Li, G.; An, T.; Gao, Y.; Fu, J. Photocatalytic degradation kinetics and mechanism of environmental pharmaceuticals in aqueous suspension of TiO2: A case of sulfa drugs. Catal. Today 2010, 153, 200–207. [Google Scholar] [CrossRef]
- Lee, Q.Y.; Li, H. Photocatalytic Degradation of Plastic Waste: A Mini Review. Micromachines 2021, 12, 907. [Google Scholar] [CrossRef]
- Nabi, I.; Bacha, A.-U.-R.; Ahmad, F.; Zhang, L. Application of titanium dioxide for the photocatalytic degradation of macro- and micro-plastics: A review. J. Environ. Chem. Eng. 2021, 9, 105964. [Google Scholar] [CrossRef]
- Žabar, R.; Komel, T.; Fabjan, J.; Kralj, M.B.; Trebše, P. Photocatalytic degradation with immobilised TiO2 of three selected neonicotinoid insecticides: Imidacloprid, thiamethoxam and clothianidin. Chemosphere 2012, 89, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Moctezuma, E.; Leyva, E.; Aguilar, C.A.; Luna, R.A.; Montalvo, C. Photocatalytic degradation of paracetamol: Intermediates and total reaction mechanism. J. Hazard. Mater. 2012, 243, 130–138. [Google Scholar] [CrossRef]
- Goutham, R.; Badri Narayan, R.; Srikanth, B.; Gopinath, K.P. Supporting Materials for Immobilisation of Nano-photocatalysts. In Nanophotocatalysis and Environmental Applications: Materials and Technology; Springer: Cham, Switzerland, 2019; pp. 49–82. [Google Scholar]
- Srikanth, B.; Goutham, R.; Badri Narayan, R.; Ramprasath, A.; Gopinath, K.P.; Sankaranarayanan, A.R. Recent advancements in supporting materials for immobilised photocatalytic applications in waste water treatment. J. Environ. Manag. 2017, 200, 60–78. [Google Scholar] [CrossRef]
- Shan, A.Y.; Ghazi, T.I.M.; Rashid, S.A. Immobilisation of titanium dioxide onto supporting materials in heterogeneous photocatalysis: A review. Appl. Catal. A Gen. 2010, 389, 1–8. [Google Scholar] [CrossRef]
- Tchinda, R.; Schwarze, M.; Schomäcker, R.; Tasbihi, M. Comparison of Commercial Nanosized Titania Particles for the Degradation of Diclofenac. J. Nanosci. Nanotechnol. 2018, 18, 7952–7959. [Google Scholar] [CrossRef]
- Schwarze, M.; Klingbeil, C.; Do, H.U.; Kutorglo, E.M.; Parapat, R.Y.; Tasbihi, M. Highly Active TiO2 Photocatalysts for Hydrogen Production through a Combination of Commercial TiO2 Material Selection and Platinum Co-Catalyst Deposition Using a Colloidal Approach with Green Reductants. Catalysts 2021, 11, 1027. [Google Scholar] [CrossRef]
- Oblak, R.; Kete, M.; Štangar, U.L.; Tasbihi, M. Alternative support materials for titania photocatalyst towards degradation of organic pollutants. J. Water Process Eng. 2018, 23, 142–150. [Google Scholar] [CrossRef]
- Moosavi, S.; Li, R.Y.M.; Lai, C.W.; Yusof, Y.; Gan, S.; Akbarzadeh, O.; Chowhury, Z.Z.; Yue, X.-G.; Johan, M.R. Methylene Blue Dye Photocatalytic Degradation over Synthesised Fe3O4/AC/TiO2 Nano-Catalyst: Degradation and Reusability Studies. Nanomaterials 2020, 10, 2360. [Google Scholar] [CrossRef]
- Muangmora, R.; Kemacheevakul, P.; Punyapalakul, P.; Chuangchote, S. Enhanced Photocatalytic Degradation of Caffeine Using Titanium Dioxide Photocatalyst Immobilized on Circular Glass Sheets under Ultraviolet C Irradiation. Catalysts 2020, 10, 964. [Google Scholar] [CrossRef]
- Trinh, D.T.T.; Le, S.T.T.; Channei, D.; Khanitchaidecha, W.; Nakaruk, A. Investigation of Intermediate Compounds of Phenol in Photocatalysis Process. Int. J. Chem. Eng. Appl. 2016, 7, 273–276. [Google Scholar] [CrossRef] [Green Version]
- Belekbir, S.; El Azzouzi, M.; El Hamidi, A.; Rodríguez-Lorenzo, L.; Santaballa, J.A.; Canle, M. Improved Photocatalyzed Degradation of Phenol, as a Model Pollutant, over Metal-Impregnated Nanosized TiO2. Nanomaterials 2020, 10, 996. [Google Scholar] [CrossRef]
- Morjène, L.; Aloulou, F.; Tasbihi, M.; Schwarze, M.; Schomäcker, R.; Seffen, M. New composite material based on Kaolinite, cement, TiO2 for efficient removal of phenol by photocatalysis. Environ. Sci. Pollut. Res. 2021, 28, 35991–36003. [Google Scholar] [CrossRef] [PubMed]
- Holm, A.; Hamandi, M.; Sahel, K.; Dappozze, F.; Guillard, C. Impact of H2O2 on the Lactic and Formic Acid Degradation in Presence of TiO2 Rutile and Anatase Phases under UV and Visible Light. Catalysts 2020, 10, 1131. [Google Scholar] [CrossRef]
- Lachheb, H.; Houas, A.; Herrmann, J.-M. Photocatalytic Degradation of Polynitrophenols on Various Commercial Suspended or Deposited Titania Catalysts Using Artificial and Solar Light. Int. J. Photoenergy 2008, 2008, 497895. [Google Scholar] [CrossRef] [Green Version]
- Ajmal, A.; Majeed, I.; Malik, R.N.; Idriss, H.; Nadeem, M.A. Principles and mechanisms of photocatalytic dye degradation on TiO2 based photocatalysts: A comparative overview. RSC Adv. 2014, 4, 37003–37026. [Google Scholar] [CrossRef]
- Fernandez, J.; Bandara, J.; Lopez, A.; Buffat, P.; Kiwi, J. Photoassisted Fenton Degradation of Nonbiodegradable Azo Dye (Orange II) in Fe-Free Solutions Mediated by Cation Transfer Membranes. Langmuir 1999, 15, 185–192. [Google Scholar] [CrossRef]
- Liu, Z.; Hosseinzadeh, S.; Wardenier, N.; Verheust, Y.; Chys, M.; Hulle, S. Van Combining ozone with UV and H2O2 for the degradation of micropollutants from different origins: Lab-scale analysis and optimization. Environ. Technol. 2019, 40, 3773–3782. [Google Scholar] [CrossRef]
- Li, W.; Jain, T.; Ishida, K.; Liu, H. A mechanistic understanding of the degradation of trace organic contaminants by UV/hydrogen peroxide, UV/persulfate and UV/free chlorine for water reuse. Environ. Sci. Water Res. Technol. 2017, 3, 128–138. [Google Scholar] [CrossRef]
- Huang, Y.; Kong, M.; Coffin, S.; Cochran, K.H.; Westerman, D.C.; Schlenk, D.; Richardson, S.D.; Lei, L.; Dionysiou, D.D. Degradation of contaminants of emerging concern by UV/H2O2 for water reuse: Kinetics, mechanisms, and cytotoxicity analysis. Water Res. 2020, 174, 115587. [Google Scholar] [CrossRef]
- Esplugas, S.; Giménez, J.; Contreras, S.; Pascual, E.; Rodríguez, M. Comparison of different advanced oxidation processes for phenol degradation. Water Res. 2002, 36, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Nosaka, Y.; Nosaka, A. Understanding Hydroxyl Radical (•OH) Generation Processes in Photocatalysis. ACS Energy Lett. 2016, 1, 356–359. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, M.F.; Michorius, A.; Buwalda, H.; Panneman, H.; Winkelman, J.G.; Beenackers, A.A.C. Comparison of the efficiency of immobilized and suspended systems in photocatalytic degradation. Catal. Today 2001, 66, 487–494. [Google Scholar] [CrossRef]
- Mascolo, G.; Comparelli, R.; Curri, M.L.; Lovecchio, G.; Lopez, A.; Agostiano, A. Photocatalytic degradation of methyl red by TiO2: Comparison of the efficiency of immobilized nanoparticles versus conventional suspended catalyst. J. Hazard. Mater. 2007, 142, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Verbruggen, S.W.; Dirckx, J.J.J.; Martens, J.A.; Lenaerts, S. Surface photovoltage measurements: A quick assessment of the photocatalytic activity? Catal. Today 2013, 209, 215–220. [Google Scholar] [CrossRef]
- Kete, M.; Pavlica, E.; Fresno, F.; Bratina, G.; Štangar, U.L. Highly active photocatalytic coatings prepared by a low-temperature method. Environ. Sci. Pollut. Res. 2014, 21, 11238–11249. [Google Scholar] [CrossRef]
- Carabin, A.; Drogui, P.; Robert, D. Photo-degradation of carbamazepine using TiO2 suspended photocatalysts. J. Taiwan Inst. Chem. Eng. 2015, 54, 109–117. [Google Scholar] [CrossRef]
- Kane, A.; Assadi, A.A.; El Jery, A.; Badawi, A.K.; Kenfoud, H.; Baaloudj, O.; Assadi, A.A. Advanced Photocatalytic Treatment of Wastewater Using Immobilized Titanium Dioxide as a Photocatalyst in a Pilot-Scale Reactor: Process Intensification. Materials 2022, 15, 4547. [Google Scholar] [CrossRef]
- Morjène, L.; Schwarze, M.; Seffen, M.; Schomäcker, R.; Tasbihi, M. Immobilization of TiO2 Semiconductor Nanoparticles onto Posidonia Oceanica Fibers for Photocatalytic Phenol Degradation. Water 2021, 13, 2948. [Google Scholar] [CrossRef]
- Sinnott, R.; Towler, G. Chemical Engineering Design; Elsevier: Amsterdam, The Netherlands, 2020; ISBN 9780081025994. [Google Scholar]
- Keane, D.A.; McGuigan, K.G.; Ibáñez, P.F.; Polo-López, M.I.; Byrne, J.A.; Dunlop, P.S.M.; O’Shea, K.; Dionysiou, D.D.; Pillai, S.C. Solar photocatalysis for water disinfection: Materials and reactor design. Catal. Sci. Technol. 2014, 4, 1211–1226. [Google Scholar] [CrossRef] [Green Version]
- Zayani, G.; Bousselmi, L.; Mhenni, F.; Ghrabi, A. Solar photocatalytic degradation of commercial textile azo dyes: Performance of pilot plant scale thin film fixed-bed reactor. Desalination 2009, 246, 344–352. [Google Scholar] [CrossRef]
- Chan, A.H.C.; Chan, C.K.; Barford, J.P.; Porter, J.F. Solar photocatalytic thin film cascade reactor for treatment of benzoic acid containing wastewater. Water Res. 2003, 37, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Joss, A.; Siegrist, H.; Ternes, T.A. Are we about to upgrade wastewater treatment for removing organic micropollutants? Water Sci. Technol. 2008, 57, 251–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchner, G.A.; Zimmermann, A.W.; Hohgräve, A.E.; Schomäcker, R. Techno-economic Assessment Framework for the Chemical Industry—Based on Technology Readiness Levels. Ind. Eng. Chem. Res. 2018, 57, 8502–8517. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photocatalyst | CS (nm) | SA (m2 g−1) | A:R | BGE (eV) | References |
|---|---|---|---|---|---|
| P25 | 18.1 | 56 | 82:18 | 3.2 | [27] |
| P90 | 12.6 | 102 | 87:13 | 3.3 | [27] |
| PC105 | 20.9 | 80 | 100:0 | 3.3 | [28] |
| PC500 | 6.0 | 270 | 100:0 | 3.3 | [28] |
| Photocatalyst | k1st, AtmO (10−3 min−1) | k1st, SynA (10−3 min−1) | k1st, H2O2 (10−3 min−1) |
|---|---|---|---|
| Photolysis | 0.5 | 0.3 | 10.6 |
| P25 | 1.7 | 3.0 | 19.0 |
| P90 | 1.4 | 2.3 | 33.9 |
| PC105 | 1.3 | 2.7 | 29.1 |
| PC500 | 0.9 | 2.2 | 7.8 |
| With Sunlight | With LED | |
|---|---|---|
| Vday (m3) | 5 | 5 |
| VYear (m3) | 1150 | 1150 |
| OpEx (EUR/Y) | 4130 | 191,600 |
| CapEx (EUR) | 656,710 | 4,657,086 |
| Treatment costs (EUR/m3) | 61 | 572 |
| Total treatment costs (EUR/Y) | 70,150 | 657,800 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarze, M.; Borchardt, S.; Frisch, M.L.; Collis, J.; Walter, C.; Menezes, P.W.; Strasser, P.; Driess, M.; Tasbihi, M. Degradation of Phenol via an Advanced Oxidation Process (AOP) with Immobilized Commercial Titanium Dioxide (TiO2) Photocatalysts. Nanomaterials 2023, 13, 1249. https://doi.org/10.3390/nano13071249
Schwarze M, Borchardt S, Frisch ML, Collis J, Walter C, Menezes PW, Strasser P, Driess M, Tasbihi M. Degradation of Phenol via an Advanced Oxidation Process (AOP) with Immobilized Commercial Titanium Dioxide (TiO2) Photocatalysts. Nanomaterials. 2023; 13(7):1249. https://doi.org/10.3390/nano13071249
Chicago/Turabian StyleSchwarze, Michael, Steffen Borchardt, Marvin L. Frisch, Jason Collis, Carsten Walter, Prashanth W. Menezes, Peter Strasser, Matthias Driess, and Minoo Tasbihi. 2023. "Degradation of Phenol via an Advanced Oxidation Process (AOP) with Immobilized Commercial Titanium Dioxide (TiO2) Photocatalysts" Nanomaterials 13, no. 7: 1249. https://doi.org/10.3390/nano13071249