
Manifesting Epoxide and Hydroxyl Groups in XPS Spectra and Valence Band of Graphene Derivatives
 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of E-xy Graphene, H-xy Graphene, and rGO
2.3. CMG Characterization
2.4. DFT Modeling
3. Results
3.1. FTIR and NMR Studies
3.2. Characterization of E-xy and H-xy Graphene via X-ray Photoelectron Spectroscopy
3.3. Probing of the E-xy and H-xy Graphene via X-ray Absorption Spectroscopy
3.4. Examination of the Valence Band of E-xy and H-xy Graphene
3.5. Modeling Spatial and Energy Distribution of E-xy and H-xy Group-Related Electron Density
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Backes, C.; Abdelkader, A.M.; Alonso, C.; Andrieux-Ledier, A.; Arenal, R.; Azpeitia, J.; Balakrishnan, N.; Banszerus, L.; Barjon, J.; Bartali, R.; et al. Production and processing of graphene and related materials. 2D Mater. 2020, 7, 022001. [Google Scholar] [CrossRef]
- Hartman, T.; Sofer, Z. Beyond Graphene: Chemistry of Group 14 Graphene Analogues: Silicene, Germanene, and Stanene. ACS Nano 2019, 13, 8566–8576. [Google Scholar] [CrossRef]
- Glavin, N.R.; Rao, R.; Varshney, V.; Bianco, E.; Apte, A.; Roy, A.; Ringe, E.; Ajayan, P.M. Emerging Applications of Elemental 2D Materials. Adv. Mat. 2020, 32, 1904302. [Google Scholar] [CrossRef]
- Zhang, C.; Gong, C.; Nie, Y.; Min, K.-A.; Liang, C.; Oh, Y.J.; Zhang, H.; Wang, W.; Hong, S.; Colombo, L.; et al. Systematic study of electronic structure and band alignment of monolayer transition metal dichalcogenides in Van der Waals heterostructures. 2D Mater. 2016, 4, 015026. [Google Scholar] [CrossRef]
- Zeng, Q.; Wang, H.; Fu, W.; Gong, Y.; Zhou, W.; Ajayan, P.M.; Lou, J.; Liu, Z. Band engineering for novel two-dimensional atomic layers. Small 2015, 11, 1868–1884. [Google Scholar] [CrossRef]
- Meng, Z.; Zhuang, J.; Xu, X.; Hao, W.; Dou, S.X.; Du, Y. Electronic Band Engineering in Elemental 2D Materials. Adv. Mater. Interfaces 2018, 5, 1800749. [Google Scholar] [CrossRef]
- Kononenko, O.; Brzhezinskaya, M.; Zotov, A.; Korepanov, V.; Levashov, V.; Matveev, V.; Roshchupkin, D. Influence of numerous Moiré superlattices on transport properties of twisted multilayer graphene. Carbon 2022, 194, 52–61. [Google Scholar] [CrossRef]
- Brzhezinskaya, M.; Kononenko, O.; Matveev, V.; Zotov, A.; Khodos, I.I.; Levashov, V.; Volkov, V.; Bozhko, S.I.; Chekmazov, S.V.; Roshchupkin, D. Engineering of Numerous Moiré Superlattices in Twisted Multilayer Graphene for Twistronics and Straintronics Applications. ACS Nano 2021, 15, 12358. [Google Scholar] [CrossRef]
- Fan, M.; Feng, Z.-Q.; Zhu, C.; Chen, X.; Chen, C.; Yang, J.; Sun, D. Recent progress in 2D or 3D N-doped graphene synthesis and the characterizations, properties, and modulations of N species. J. Mater. Sci. 2016, 51, 10323–10349. [Google Scholar] [CrossRef]
- Hirsch, A.; Hauke, F. Post-Graphene 2D Chemistry: The Emerging Field of Molybdenum Disulfide and Black Phosphorus Functionalization. Angew. Chem. Int. Ed. 2018, 57, 4338–4354. [Google Scholar] [CrossRef]
- Yu, W.; Sisi, L.; Haiyan, Y.; Jie, L. Progress in the functional modification of graphene/graphene oxide: A review. RSC Adv. 2020, 10, 15328–15345. [Google Scholar] [CrossRef]
- Pumera, M.; Sofer, Z. Towards stoichiometric analogues of graphene: Graphane, fluorographene, graphol, graphene acid and others. Chem. Soc. Rev. 2017, 46, 4450–4463. [Google Scholar] [CrossRef] [Green Version]
- Matochová, D.; Medved’, M.; Bakandritsos, A.; Steklý, T.; Zbořil, R.; Otyepka, M. 2D Chemistry: Chemical Control of Graphene Derivatization. J. Phys. Chem. Lett. 2018, 9, 3580–3585. [Google Scholar] [CrossRef]
- Brisebois, P.P.; Siaj, M. Harvesting graphene oxide—Years 1859 to 2019: A review of its structure, synthesis, properties and exfoliation. J. Mater. Chem. C 2020, 8, 1517–1547. [Google Scholar] [CrossRef]
- Javaid, S.; Akhtar, M.J.; Kim, E.M.; Lee, G. Band gap tuning and excitonic effect in chloro-fluorinated graphene. Surf. Sci. 2019, 686, 39–44. [Google Scholar] [CrossRef]
- Jeong, H.K.; Yang, C.; Kim, B.S.; Kim, K.-j. Valence band of graphite oxide. Europhys. Lett. 2010, 92, 37005. [Google Scholar] [CrossRef]
- Sutar, D.S.; Singh, G.; Botcha, V.D. Electronic structure of graphene oxide and reduced graphene oxide monolayers. Appl. Phys. Lett. 2012, 101, 103103. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Ogawa, S.; Watanabe, D.; Hozumi, H.; Gao, Y.; Eda, G.; Mattevi, C.; Fujita, T.; Yoshigoe, A.; Ishizuka, S.; et al. Valence-band electronic structure evolution of graphene oxide upon thermal annealing for optoelectronics. Phys. Status Solidi A 2016, 213, 2380–2386. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Zheng, Y.; Podkolzin, S.G.; Lee, W. Band gap of reduced graphene oxide tuned by controlling functional groups. J. Mater. Chem. C 2020, 8, 4885–4894. [Google Scholar] [CrossRef]
- Rabchinskii, M.K.; Ryzhkov, S.A.; Gudkov, M.V.; Baidakova, M.V.; Saveliev, S.D.; Pavlov, S.I.; Shnitov, V.V.; Kirilenko, D.A.; Stolyarova, D.Y.; Lebedev, A.M.; et al. Unveiling a facile approach for large-scale synthesis of N-doped graphene with tuned electrical properties. 2D Mater. 2020, 7, 045001. [Google Scholar] [CrossRef]
- Chang, C.-K.; Kataria, S.; Kuo, C.-C.; Ganguly, A.; Wang, B.-Y.; Hwang, J.-Y.; Huang, K.-J.; Yang, W.-H.; Wang, S.-B.; Chuang, C.-H.; et al. Band Gap Engineering of Chemical Vapor Deposited Graphene by in Situ BN Doping. ACS Nano 2013, 7, 1333–1341. [Google Scholar] [CrossRef]
- Yang, C.-S.; Han, Y.; Ye, Y.; Pan, H.; Lee, J.-O.; Zhu, J.; Jeong, H.K. Valence band of poly(sodium 4-styrenesulfonate) intercalated graphite oxide composites. Chem. Phys. Lett. 2013, 559, 67–70. [Google Scholar] [CrossRef]
- Favaro, M.; Rizzi, G.A.; Nappini, S.; Magnano, E.; Bondino, F.; Agnoli, S.; Granozzi, G. A synchrotron-based spectroscopic study of the electronic structure of N-doped HOPG and PdY/N-doped HOPG. Surf. Sci. 2016, 646, 132–139. [Google Scholar] [CrossRef]
- Tang, L.; Meng, X.; Deng, D.; Bao, X. Confinement Catalysis with 2D Materials for Energy Conversion. Adv. Mat. 2019, 31, 1901996. [Google Scholar] [CrossRef]
- Tan, W.C.; Ang, K.-W. Volatile Organic Compound Sensors Based on 2D Materials. Adv. Electron. Mater. 2021, 7, 2001071. [Google Scholar] [CrossRef]
- Mathew, M.; Shinde, P.V.; Samal, R.; Rout, C.S. A review on mechanisms and recent developments in p-n heterojunctions of 2D materials for gas sensing applications. J. Mater. Sci. 2021, 56, 9575–9604. [Google Scholar] [CrossRef]
- Wang, Y.; Mao, J.; Meng, X.; Yu, L.; Deng, D.; Bao, X. Catalysis with Two-Dimensional Materials Confining Single Atoms: Concept, Design, and Applications. Chem. Rev. 2019, 119, 1806–1854. [Google Scholar] [CrossRef]
- Chaurasiya, R.; Dixit, A. Defect engineered MoSSe Janus monolayer as a promising two dimensional material for NO2 and NO gas sensing. Appl. Surf. Sci. 2019, 490, 204–219. [Google Scholar] [CrossRef] [Green Version]
- Tit, N.; Said, K.; Mahmoud, N.M.; Kouser, S.; Yamani, Z.H. Ab-initio investigation of adsorption of CO and CO2 molecules on graphene: Role of intrinsic defects on gas sensing. Appl. Surf. Sci. 2017, 394, 219–230. [Google Scholar] [CrossRef]
- Zhang, Z.; Cai, R.; Long, F.; Wang, J. Development and application of tetrabromobisphenol A imprinted electrochemical sensor based on graphene/carbon nanotubes three-dimensional nanocomposites modified carbon electrode. Talanta 2015, 134, 435–442. [Google Scholar] [CrossRef]
- Shangguan, Q.; Chen, Z.; Yang, H.; Cheng, S.; Yang, W.; Yi, Z.; Wu, X.; Wang, S.; Yi, Y.; Wu, P. Design of Ultra-Narrow Band Graphene Refractive Index Sensor. Sensors 2022, 22, 6483. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, Z.; Yang, H.; Wenc, L.; Yi, Z.; Zhou, Z.; Dai, B.; Zhang, J.; Wue, X.; Wu, P. Multi-mode surface plasmon resonance absorber based on dart-type single-layer graphene. RSC Adv. 2022, 12, 7821–7829. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Liao, J.; He, B.; Zhang, F.; Zhang, F.; Huang, X.; Zhou, L. One-Step Fabrication of Graphene Oxide Enhanced Magnetic Composite Gel for Highly Efficient Dye Adsorption and Catalysis. ACS Sustain. Chem. Eng. 2015, 3, 1677–1685. [Google Scholar] [CrossRef]
- Shnitov, V.V.; Rabchinskii, M.K.; Brzhezinskaya, M.; Stolyarova, D.Y.; Pavlov, S.V.; Baidakova, M.V.; Shvidchenko, A.V.; Kislenko, V.A.; Kislenko, S.A.; Brunkov, P.N. Valence Band Structure Engineering in Graphene Derivatives. Small 2021, 17, 2104316. [Google Scholar] [CrossRef] [PubMed]
- Rabchinskii, M.K.; Sysoev, V.V.; Ryzhkov, S.A.; Eliseyev, I.A.; Stolyarova, D.Y.; Antonov, G.A.; Struchkov, N.S.; Brzhezinskaya, M.; Kirilenko, D.A.; Pavlov, S.I.; et al. A blueprint for the synthesis and characterization of the thiolated graphene. Nanomaterials 2022, 12, 45. [Google Scholar] [CrossRef]
- Sun, J.; Deng, Y.; Li, J.; Wang, G.; He, P.; Tian, S.; Bu, X.; Di, Z.; Yang, S.; Ding, G.; et al. A New Graphene Derivative: Hydroxylated Graphene with Excellent Biocompatibility. ACS Appl. Mater. Interfaces 2016, 8, 10226–10233. [Google Scholar] [CrossRef]
- Vacchi, I.A.; Raya, J.; Bianco, A.; Ménard-Moyon, C. Controlled derivatization of hydroxyl groups of graphene oxide in mild conditions. 2D Mater. 2018, 5, 035037. [Google Scholar] [CrossRef]
- Dimiev, A.; Kosynkin, D.V.; Alemany, L.B.; Chaguine, P.; Tour, J.M. Pristine Graphite Oxide. J. Am. Chem. Soc. 2012, 134, 2815–2822. [Google Scholar] [CrossRef]
- Molodtsov, S.L.; Fedoseenko, S.I.; Vyalikh, D.V.; Iossifov, I.E.; Follath, R.; Gorovikov, S.A.; Brzhezinskaya, M.M.; Dedkov, Y.S.; Püttner, R.; Schmidt, J.-S.; et al. High-resolution Russian–German beamline at BESSY. Appl. Phys. A 2009, 94, 501–505. [Google Scholar] [CrossRef]
- Sakhonenkov, S.S.; Filatova, E.O.; Gaisin, A.U.; Kasatikov, S.A.; Konashuk, A.S.; Pleshkov, R.S.; Chkhalo, N.I. Angle resolved photoelectron spectroscopy as applied to X-ray mirrors: An in depth study of Mo/Si multilayer systems. Phys. Chem. Chem. Phys. 2019, 21, 25002–25010. [Google Scholar] [CrossRef]
- Stöhr, J. NEXAFS Spectroscopy, Springer Series in Surface Sciences; Springer: New York, NY, USA, 1992; ISBN 978-3-642-08113-2. [Google Scholar]
- Savin, G.; Shabanov, B.; Telegin, P.; Baranov, A. Joint supercomputer center of the Russian Academy of Sciences: Present and future. Lobachevskii J. Math. 2019, 40, 1853–1862. [Google Scholar] [CrossRef]
- Zacharov, I.; Arslanov, R.; Gunin, M.; Stefonishin, D.; Bykov, A.; Pavlov, S.; Panarin, O.; Maliutin, A.; Rykovanov, S.; Fedorov, M. Zhores petaflops supercomputer for data-driven modeling, machine learning and artificial intelligence installed in Skolkovo Institute of Science and Technology. Open Eng. 2019, 9, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Ţucureanu, V.; Matei, A.; Avram, A.M. FTIR Spectroscopy for Carbon Family Study. Crit. Rev. Anal. Chem. 2016, 46, 502–520. [Google Scholar] [CrossRef]
- Rabchinskii, M.K.; Ryzhkov, S.A.; Besedina, N.A.; Brzhezinskaya, M.; Malkov, M.N.; Stolyarova, D.Y.; Arutyunyan, A.F.; Struchkov, N.S.; Saveliev, S.D.; Diankin, I.D.; et al. Guiding graphene derivatization for covalent immobilization of aptamers. Carbon 2022, 196, 264–279. [Google Scholar] [CrossRef]
- Acik, M.; Lee, G.; Mattevi, C.; Pirkle, A.; Wallace, R.M.; Chhowalla, M.; Cho, K.; Chabal, Y. The Role of Oxygen during Thermal Reduction of Graphene Oxide Studied by Infrared Absorption Spectroscopy. J. Phys. Chem. C 2011, 115, 19761–19781. [Google Scholar] [CrossRef]
- Acik, M.; Lee, G.; Mattevi, C.; Chhowalla, M.; Cho, K.; Chabal, Y.J. Unusual infrared-absorption mechanism in thermally reduced graphene oxide. Nat. Mat. 2010, 9, 840–845. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef]
- Rabchinskii, M.K.; Shnitov, V.V.; Dideikin, A.T.; Aleksenskii, A.E.; Vul’, S.P.; Baidakova, M.V.; Pronin, I.I.; Kirilenko, D.A.; Brunkov, P.N.; Weise, J.; et al. Nanoscale perforation of graphene oxide during photoreduction process in the argon atmosphere. J. Phys. Chem. C 2016, 120, 28261–28269. [Google Scholar] [CrossRef]
- Ganguly, A.; Sharma, S.; Papakonstantinou, P.; Hamilton, J. Probing the Thermal Deoxygenation of Graphene Oxide Using High-Resolution in situ X-Ray-Based Spectroscopies. J. Phys. Chem. C 2011, 115, 17009–17019. [Google Scholar] [CrossRef] [Green Version]
- Rabchinskii, M.K.; Sysoev, V.V.; Glukhova, O.E.; Brzhezinskaya, M.; Stolyarova, D.Y.; Varezhnikov, A.S.; Solomatin, M.A.; Barkov, P.V.; Kirilenko, D.A.; Pavlov, S.I.; et al. Guding graphene Derivatization for the On-Chip Multisensor Arrays: From the Synthesis to the Theoretical Background. Adv. Mater. Technol. 2022, 7, 2101250. [Google Scholar] [CrossRef]
- Susi, T.; Kaukonen, M.; Havu, P.; Ljungberg, M.P.; Ayala, P.; Kauppinen, E.I. Core level binding energies of functionalized and defective graphene. Beilstein J. Nanotechnol. 2014, 5, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.Z.; Johns, J.E.; Bevan, K.H.; Karmel, H.J.; Liang, Y.T.; Yoshimoto, S.; Mukai, K.; Koitaya, T.; Yoshinobu, J.; Kawai, M.; et al. An ab initio study of oxygen on strained graphene. Nat. Chem. 2012, 4, 305–309. [Google Scholar] [CrossRef]
- Barinov, A.; Malcioglu, O.B.; Fabris, S.; Sun, T.; Gregoratti, L.; Dalmiglio, M.; Kiskinova, M.J. Core level binding energies of functionalized and defective graphene. Phys. Chem. C 2009, 113, 9009–9013. [Google Scholar] [CrossRef]
- Rabchinskii, M.K.; Shnitov, V.V.; Stolyarova, D.Y.; Ryzhkov, S.A.; Baidakova, M.V.; Lobanova, E.Y.; Shvidchenko, A.V.; Besedina, N.A.; Smirnov, D.A. Graphene oxide conversion into controllably carboxylated graphene layers via photoreduction process in the inert atmosphere. Fuller. Nanotub. Carbon Nanostruct. 2020, 28, 221–225. [Google Scholar] [CrossRef]
- Lee, V.; Dennis, R.V.; Schultz, B.J.; Jaye, C.; Fischer, D.A.; Banerjee, S.J. Soft X-ray Absorption Spectroscopy Studies of the Electronic Structure Recovery of Graphene Oxide upon Chemical Defunctionalization. Phys. Chem. C 2012, 116, 20591–20599. [Google Scholar] [CrossRef]
- Brzhezinskaya, M.; Kapitanova, O.; Kononenko, O.; Koveshnikov, S.; Korepanov, V.; Roshchupkin, D. Large-scalable graphene oxide films with resistive switching for non-volatile memory applications. J. Alloys Compd. 2020, 849, 156699. [Google Scholar] [CrossRef]
- Chuang, C.-H.; Wang, Y.-F.; Shao, Y.-C.; Yeh, Y.-C.; Wang, D.-Y.; Chen, C.-W.; Chiou, J.W.; Ray, S.C.; Pong, W.F.; Zhang, L.; et al. The Effect of Thermal Reduction on the Photoluminescence and Electronic Structures of Graphene Oxides. Sci.Rep. 2014, 4, 4525. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Wang, Y.; Zheng, H.; Wu, Z. X-ray absorption spectroscopy study on the thermal and hydrazine reduction of graphene oxide. J. Electron. Spectrosc. Relat. Phenom. 2014, 196, 89–93. [Google Scholar] [CrossRef]
- Ng, M.L.; Balog, R.; Hornekær, L.; Preobrajenski, A.B.; Vinogradov, N.A.; Mårtensson, N.; Schulte, K. Controlling Hydrogenation of Graphene on Transition Metals. J. Phys. Chem. C 2010, 114, 18559–18565. [Google Scholar] [CrossRef] [Green Version]
- Krimm, S.; Liang, C.Y.; Sutherland, G.B.B.M. Infrared Spectra of High Polymers. II. Polyethylene. J. Chem. Phys. 1956, 25, 549. [Google Scholar] [CrossRef] [Green Version]
- Kaznacheyev, K.; Osanna, A.; Jacobesen, C.; Plashkevych, O.; Vahtras, O.; Agren, H.; Carravetta, V.; Hitchcock, A.P. Innershell Absorption Spectroscopy of Amino Acids. J. Phys. Chem. A 2002, 106, 3153. [Google Scholar] [CrossRef]
- Edwards, D.C.; Myneni, S.C.B. Near Edge X-ray Absorption Fine Structure Spectroscopy of Bacterial Hydroxamate Siderophores in Aqueous Solutions. J. Phys. Chem. A 2006, 110, 11809–11818. [Google Scholar] [CrossRef] [PubMed]
- McFeely, F.R.; Kowalczyk, S.P.; Ley, L.; Cavell, R.G.; Pollak, R.A.; Shirley, D.A. X-ray photoemission studies of diamond, graphite, and glassy carbon valence bands. Phys. Rev. B 1974, 9, 5268–5278. [Google Scholar] [CrossRef] [Green Version]
- Yeh, J.J.; Lindau, I. Atomic subshell photoionization cross sections and asymmetry parameters: 1 ⩽ Z ⩽ 103. At. Data Nucl. Data Tables 1985, 32, 1–155. [Google Scholar] [CrossRef]
- Srivastava, R.; Al-Omary, F.A.M.; El-Emam, A.A.; Pathak, S.K.; Karabacak, M.; Narayan, V.; Chand, S.; Prasad, O.; Sinh, L. A combined experimental and theoretical DFT (B3LYP, CAM-B3LYP and M06-2X) study on electronic structure, hydrogen bonding, solvent effects and spectral features of methyl 1H-indol-5-carboxylate. J. Mol. Struct. 2017, 1137, 725. [Google Scholar] [CrossRef]







{kind=link}
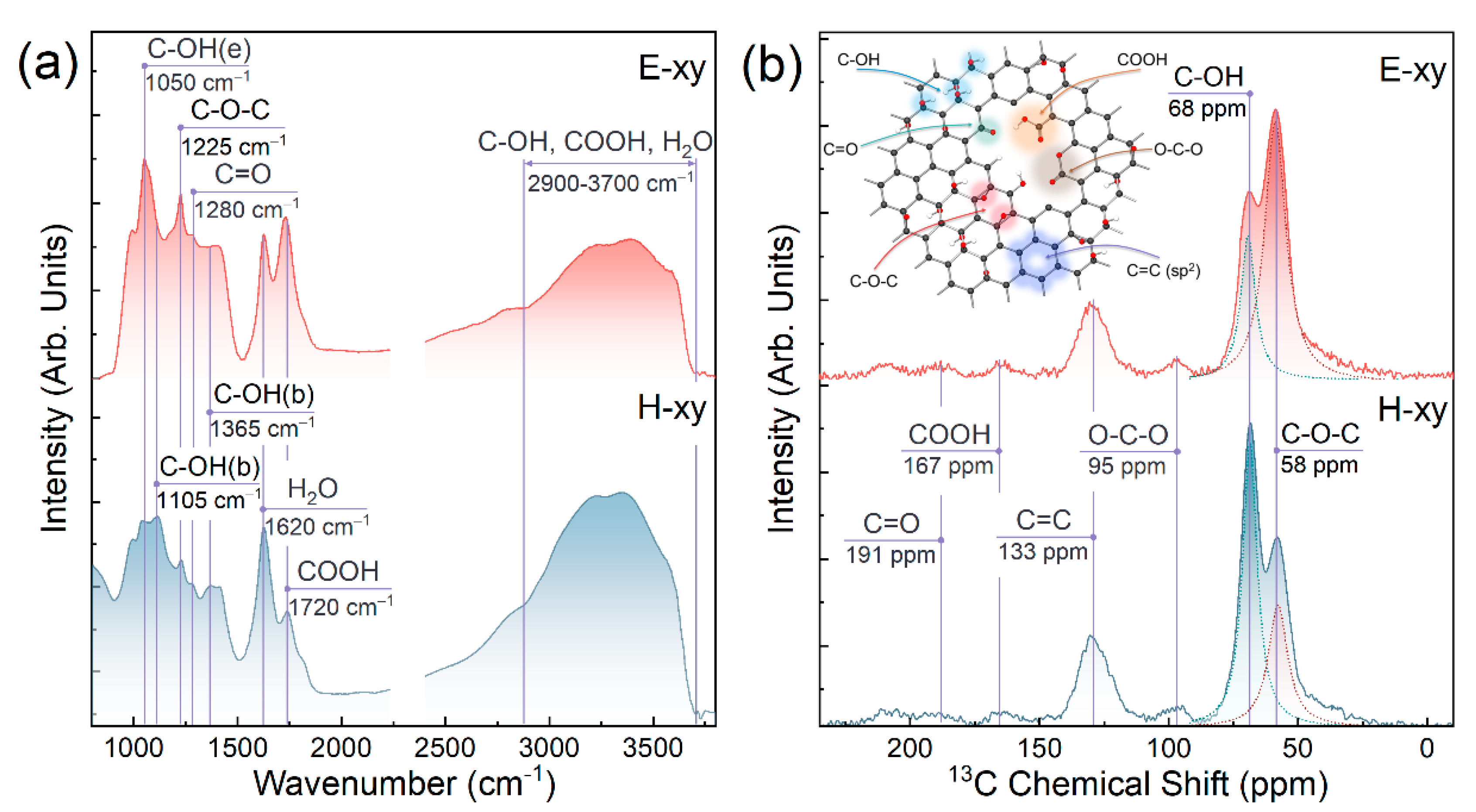{kind=link}
{kind=link}
{kind=link}
{kind=link}
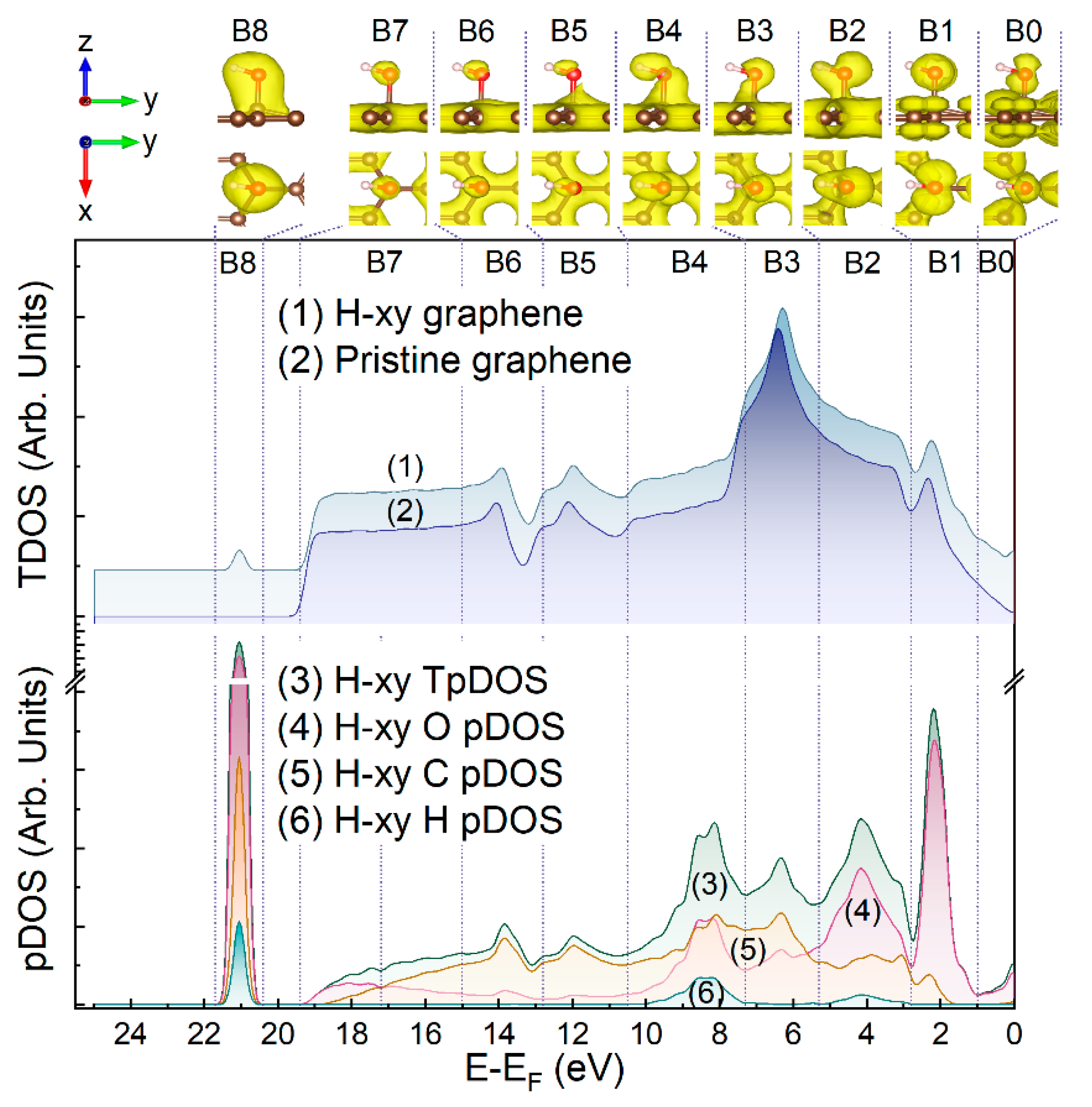{kind=link}
| Component | C=C | C-OH | C-O-C | C=O | COOH | C-O-C/C-OH Ratio |
|---|---|---|---|---|---|---|
| Binding Energy (eV) | 284.6 | 286.8 | 287.0 | 288.2 | 288.9 | |
| E-xy | 72.4 | 3.7 | 19.5 | 2.4 | 2.0 | 70%/30% |
| H-xy | 69.4 | 21.7 | 3.2 | 2.8 | 2.5 | 29%/71% |
| VB Feature | E−EF (eV) | Assignment | |||||
|---|---|---|---|---|---|---|---|
| This Work | [16] | [17] | [18] | [23] | [66] | ||
| E-xy and H-xy Graphene | GO/Graphite | GO/rGO | GO/rGO | HOPG | Graphene | ||
| X | 3.2 | (π) (C=C) | (π) (C=C) | (π) (C=C) | - | (π) (C=C) | (π) (C=C) |
| A | 5.0 | n’(O2p) | (σ) (C=C) | - | - | - | - |
| B | 7.8 | σ(C-O) | & O2p-states | - | - | (C=C) | - |
| C’ | 9.5 | (σ) (C=C) | (σ) (C=C) | - | - | - | |
| C | 10.6 | σ(C-O)/σ(O-H) | - | - | - | ||
| D | 13.5 | (σ) (C=C) | (σ) (C=C) | (σ) (C=C) | (σ) (C=C) | (C=C) | (σ) (C=C) |
| E | 17.0 | (σ) (C=C) | (σ) (C=C) | (σ) (C=C) | (σ) (C=C) | - | (σ) (C=C) |
| F | 19.3 | (σ) (C=C) | (σ) (C=C) | (σ) (C=C) | (σ) (C=C) | - | (σ)(C=C) |
| F’ | 21.5 | (σ) (C=C) | (σ) (C=C) | (σ) (C=C) | - | (σ) (C=C) | |
| G | 24.7 | O2s AOs of σ(C-O) | O2s states | - | - | - | - |
| G’ | 26.9 | O2s AOs of σ(COOH) | - | - | - | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabchinskii, M.K.; Shnitov, V.V.; Brzhezinskaya, M.; Baidakova, M.V.; Stolyarova, D.Y.; Ryzhkov, S.A.; Saveliev, S.D.; Shvidchenko, A.V.; Nefedov, D.Y.; Antonenko, A.O.; et al. Manifesting Epoxide and Hydroxyl Groups in XPS Spectra and Valence Band of Graphene Derivatives. Nanomaterials 2023, 13, 23. https://doi.org/10.3390/nano13010023
Rabchinskii MK, Shnitov VV, Brzhezinskaya M, Baidakova MV, Stolyarova DY, Ryzhkov SA, Saveliev SD, Shvidchenko AV, Nefedov DY, Antonenko AO, et al. Manifesting Epoxide and Hydroxyl Groups in XPS Spectra and Valence Band of Graphene Derivatives. Nanomaterials. 2023; 13(1):23. https://doi.org/10.3390/nano13010023
Chicago/Turabian StyleRabchinskii, Maxim K., Vladimir V. Shnitov, Maria Brzhezinskaya, Marina V. Baidakova, Dina Yu. Stolyarova, Sergey A. Ryzhkov, Svyatoslav D. Saveliev, Alexander V. Shvidchenko, Denis Yu. Nefedov, Anastasiia O. Antonenko, and et al. 2023. "Manifesting Epoxide and Hydroxyl Groups in XPS Spectra and Valence Band of Graphene Derivatives" Nanomaterials 13, no. 1: 23. https://doi.org/10.3390/nano13010023