
Band Gap Engineering of Newly Discovered ZnO/ZnS Polytypic Nanomaterials

Abstract
:1. Introduction
2. Materials and Methods
3. Results
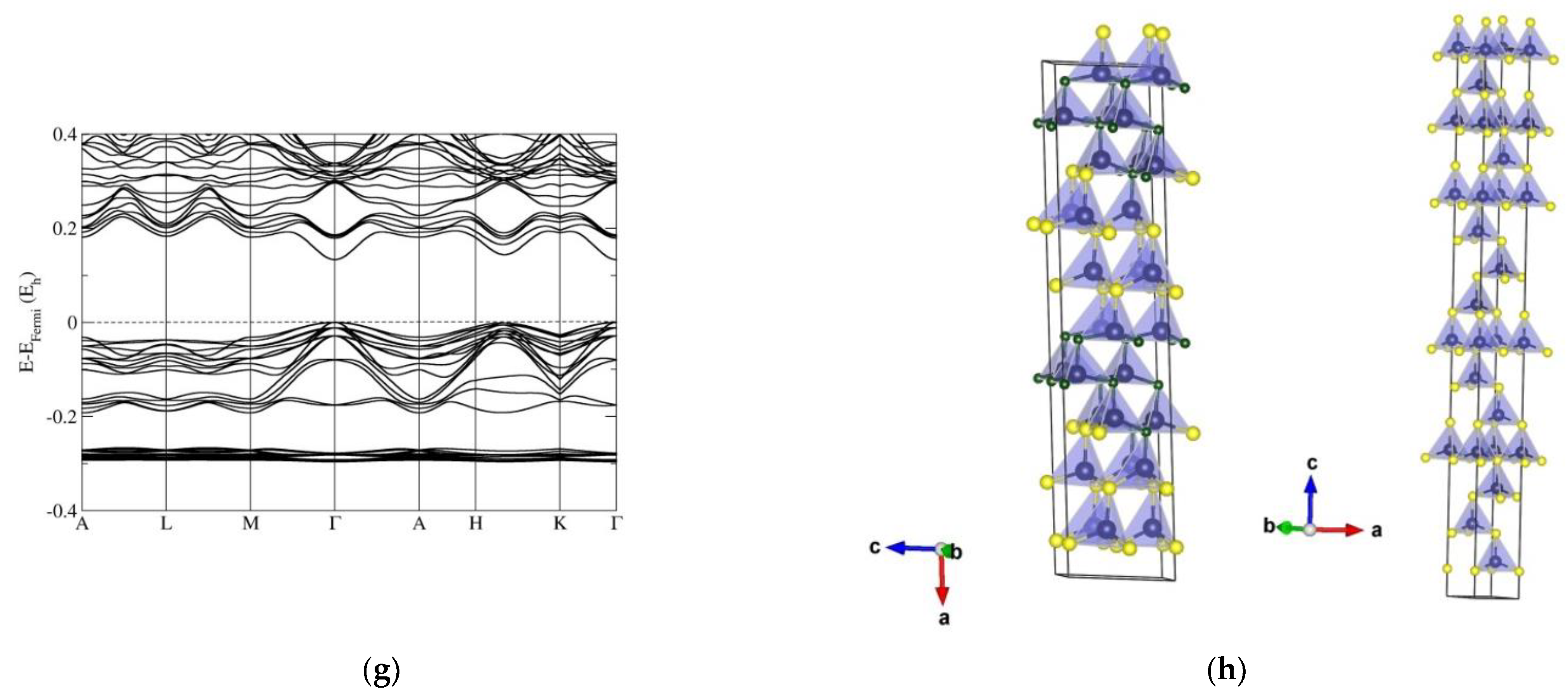
3.1. Structural Investigation of ZnO/ZnS
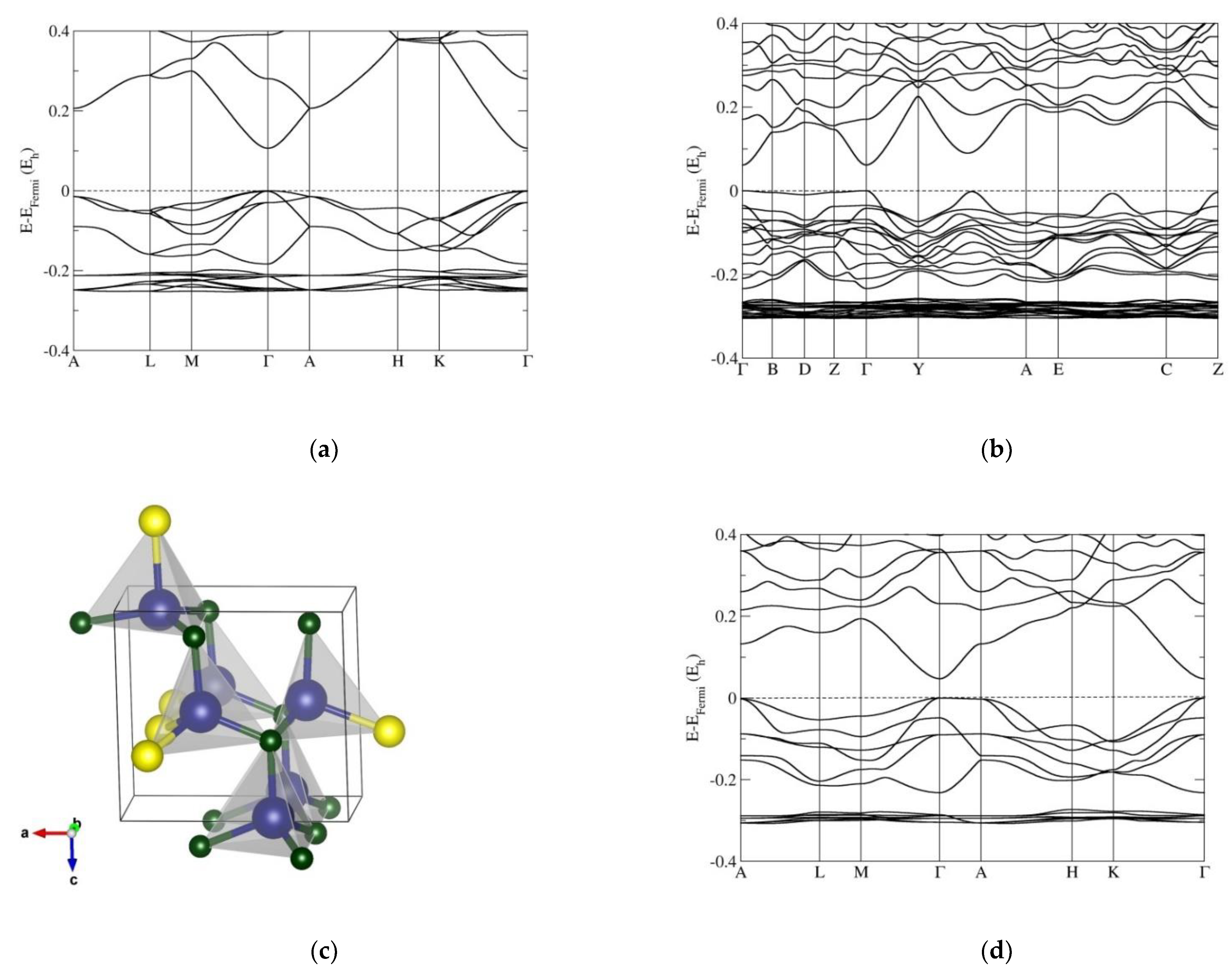
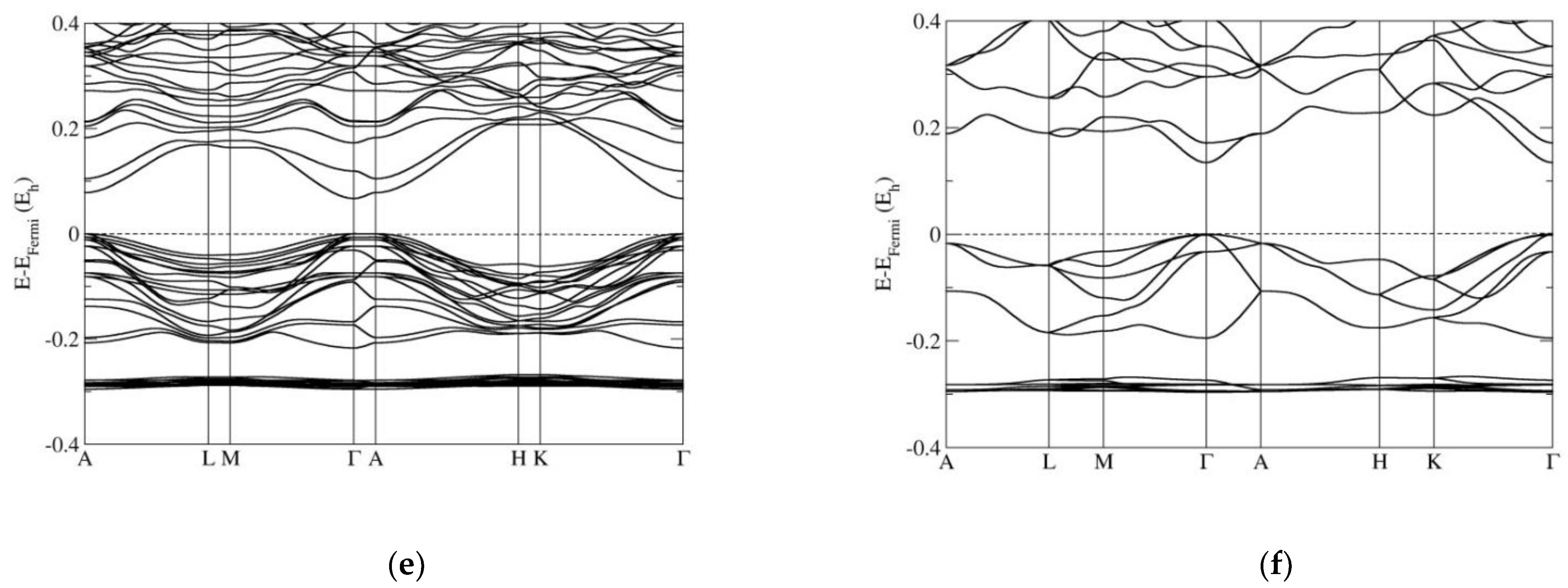
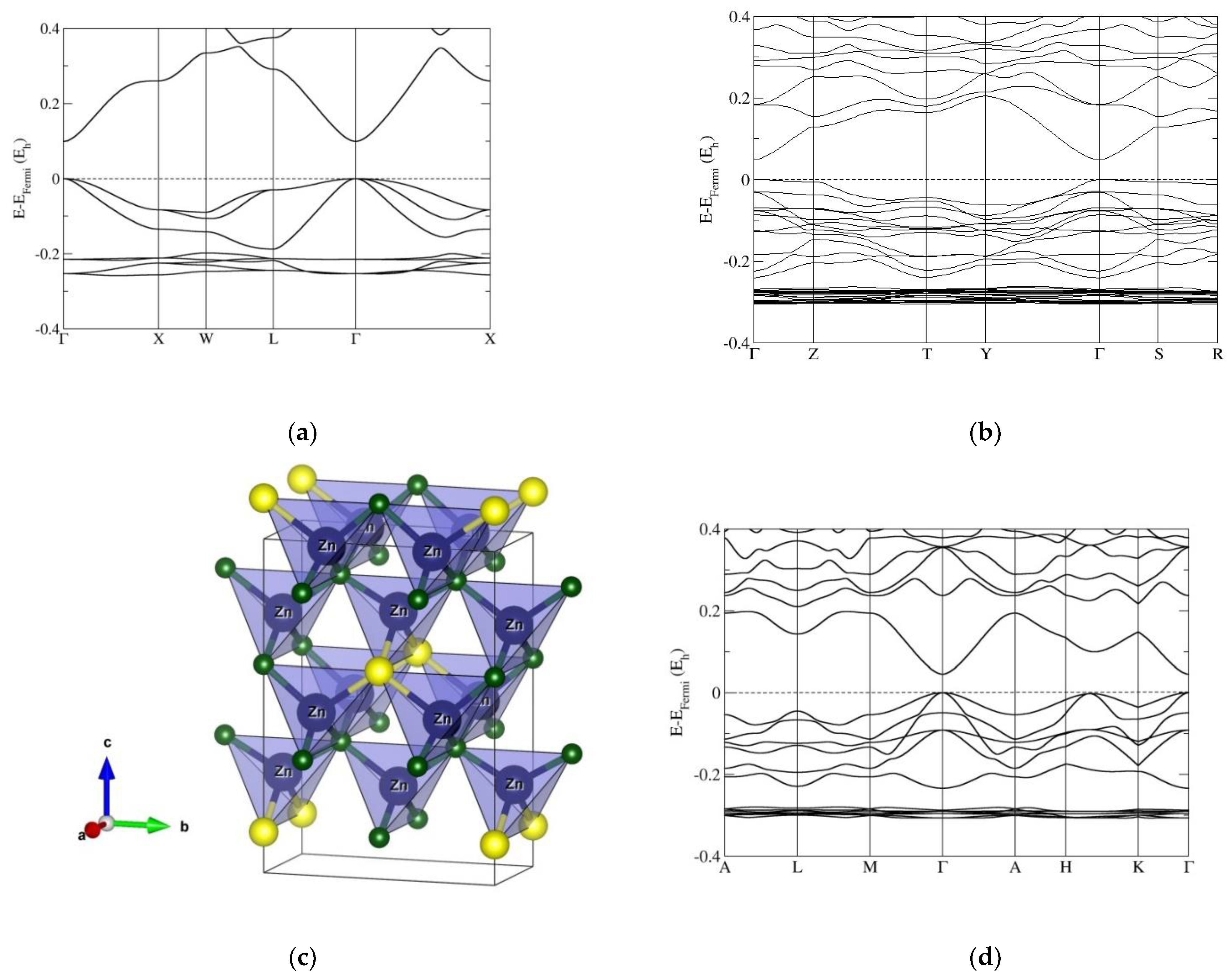
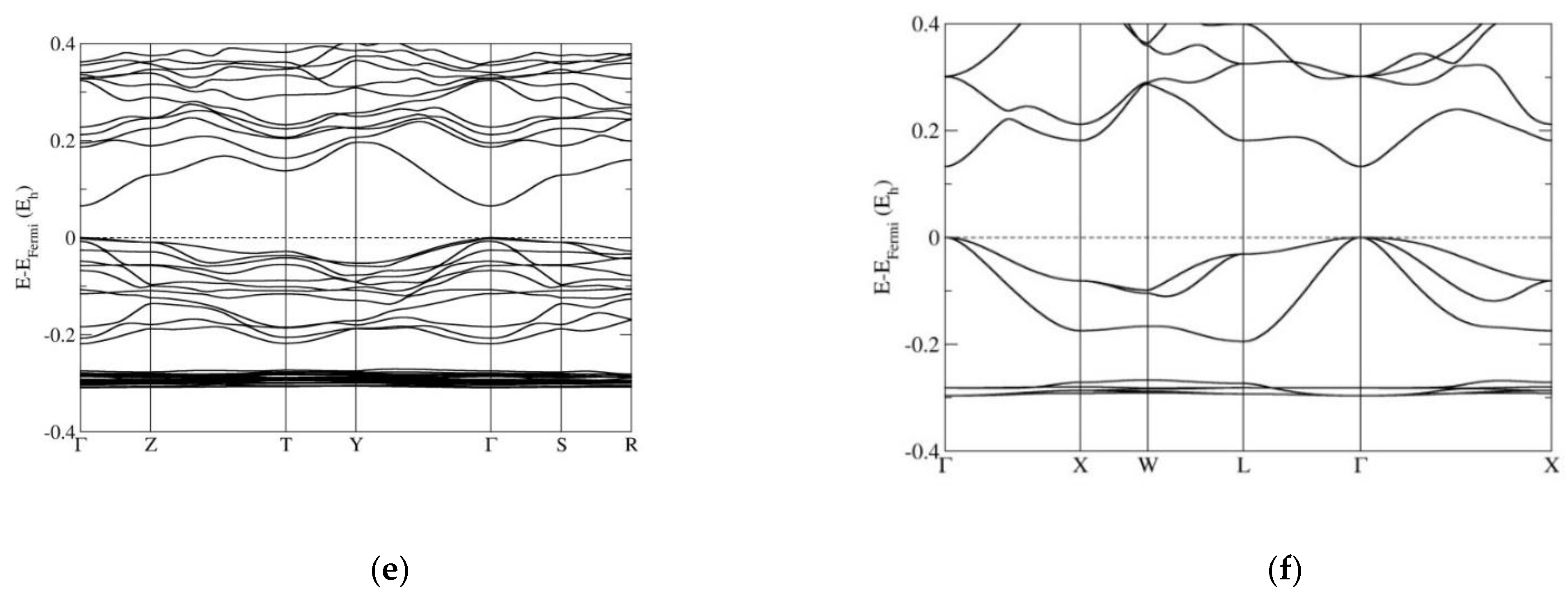
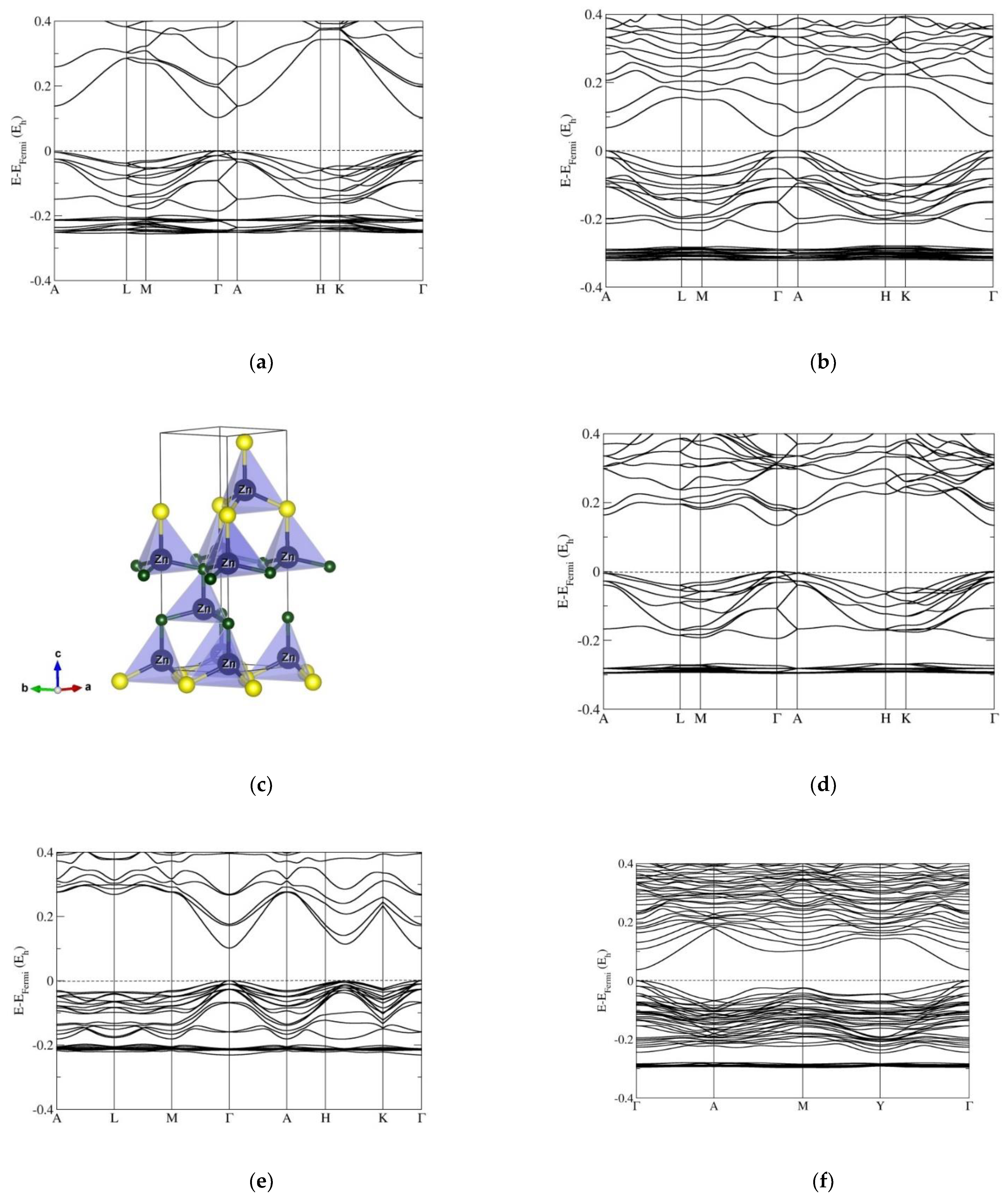
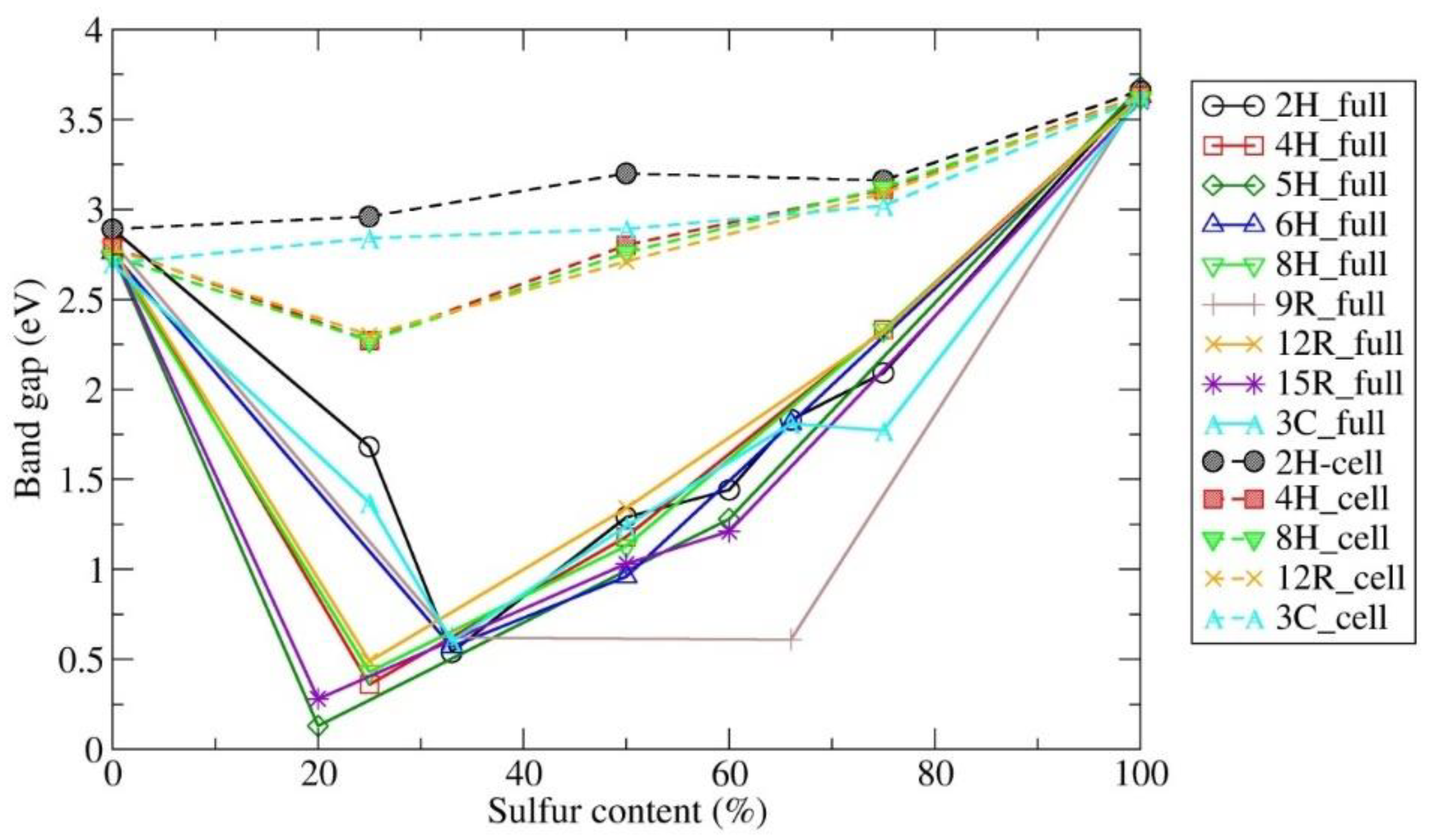
3.2. Electronic Band Structure Calculations
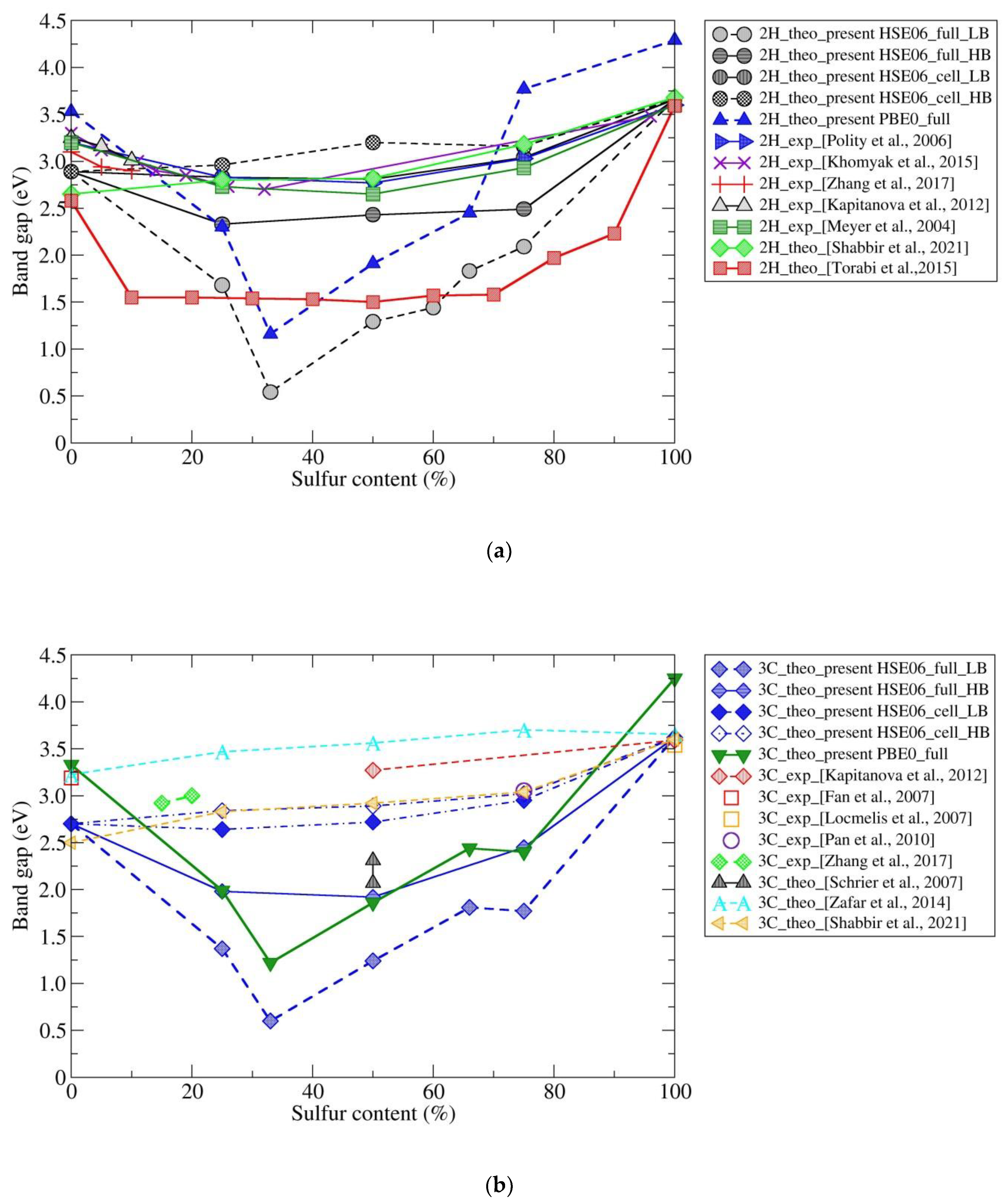
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palache, C.; Berman, H.; Frondel, C. The System of Mineralogy of James Dwight Dana and Edward Salisbury Dana Yale University 1837–1892, 7th ed.; John Wiley and Sons, Inc.: New York, NY, USA, 1944; Volume 1, p. 844. [Google Scholar]
- Anthony, J.W.; Bideaux, R.A.; Bladh, K.W.; Nichols, M.C. Handbook of Mineralogy; Mineralogical Society of America: Chantilly, VA, USA, 2022. [Google Scholar]
- Muntyan, B.L. Colorado Sphalerite. Rocks Miner. 1999, 74, 220–235. [Google Scholar] [CrossRef]
- Frondel, C.; Baum, J.L. Structure and Mineralogy of the Franklin Zinc-Iron-Manganese Deposit, New Jersey. Econ. Geol. 1974, 69, 157–180. [Google Scholar] [CrossRef]
- Gerlach, W. Das Ka-Dublett, nebst einer Neubestimmung der Gitterkonstanten einiger Krystalle. Z. Für Phys. 1922, 23, 114–120. [Google Scholar]
- Scott, S.D.; Barnes, H.L. Sphalerite-wurtzite equilibria and stoichiometry. Geochim. Cosmochim. Acta 1972, 36, 1275–1295. [Google Scholar] [CrossRef]
- Logar, M.; Jančar, B.; Rečnik, A.; Suvorov, D. Controlled synthesis of pure and doped ZnS nanoparticles in weak polyion assemblies: Growth characteristics and fluorescence properties. Nanotechnology 2009, 20, 275601. [Google Scholar] [CrossRef]
- Özgür, Ü.; Alivov, Y.I.; Liu, C.; Teke, A.; Reshchikov, M.A.; Doğan, S.; Avrutin, V.; Cho, J.H.; Morkoç, H. A comprehensive review of ZnO materials and devices. J. Appl. Phys. 2005, 98, 041301. [Google Scholar] [CrossRef] [Green Version]
- Luković Golić, D.; Branković, G.; Počuča Nešić, M.; Vojisavljević, K.; Rečnik, A.; Daneu, N.; Bernik, S.; Šćepanović, M.; Poleti, D.; Branković, Z. Structural characterization of self-assembled ZnO nanoparticles obtained by the sol–gel method from Zn(CH3 COO)2·2H2O. Nanotechnology 2011, 22, 395603. [Google Scholar] [CrossRef]
- Catlow, C.R.A.; French, S.A.; Sokol, A.A.; Al-Sunaidi Abdullah, A.; Woodley, S.M. Zinc oxide: A case study in contemporary computational solid state chemistry. J. Comput. Chem. 2008, 29, 2234–2249. [Google Scholar] [CrossRef]
- Zagorac, D.; Schön, J.C. Energy landscapes of pure and doped ZnO: From bulk crystals to nanostructures. In Energy Landscapes of Nanoscale Systems; Wales, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Rkhioui, A.; Masrour, R.; Hlil, E.K.; Bahmad, L.; Hamedoun, M.; Benyoussef, A. Study of Electronic and Magnetic Properties of Zn1−xMxO (M = Mn and Cr) by ab initio Calculations. J. Supercond. Nov. Magn. 2013, 26, 3469–3474. [Google Scholar] [CrossRef]
- Misra, P.; Sahoo, P.K.; Tripathi, P.; Kulkarni, V.N.; Nandedkar, R.V.; Kukreja, L.M. Sequential pulsed laser deposition of CdxZn1−xO alloy thin films for engineering ZnO band gap. Appl. Phys. A 2004, 78, 37–40. [Google Scholar] [CrossRef]
- Nafees, M.; Liaqut, W.; Ali, S.; Shafique, M.A. Synthesis of ZnO/Al:ZnO nanomaterial: Structural and band gap variation in ZnO nanomaterial by Al doping. Appl. Nanosci. 2013, 3, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Teehan, S.; Efstathiadis, H.; Haldar, P. Thermoelectric Performance of ZnO:Al/ZnO:(Al,In) Quantum Well Multilayer Structures as a Function of Indium Composition and Band-Gap Offset at High Operating Temperatures. J. Electron. Mater. 2012, 41, 1831–1837. [Google Scholar] [CrossRef]
- Shet, S.; Ahn, K.-S.; Deutsch, T.; Wang, H.; Ravindra, N.; Yan, Y.; Turner, J.; Al-Jassim, M. Synthesis and characterization of band gap-reduced ZnO:N and ZnO:(Al,N) films for photoelectrochemical water splitting. J. Mater. Res. 2010, 25, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Ribić, V.; Rečnik, A.; Komelj, M.; Kokalj, A.; Branković, Z.; Zlatović, M.; Branković, G. New inversion boundary structure in Sb-doped ZnO predicted by DFT calculations and confirmed by experimental HRTEM. Acta Mater. 2020, 199, 633–648. [Google Scholar] [CrossRef]
- Djurišić, A.B.; Leung, Y.H. Optical properties of ZnO nanostructures. Small 2006, 2, 944–961. [Google Scholar] [CrossRef] [PubMed]
- Janotti, A.; Van de Walle, C.G. Fundamentals of zinc oxide as a semiconductor. Rep. Prog. Phys. 2009, 72, 126501. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Bando, Y.; Gautam, U.K.; Zhai, T.; Zeng, H.; Xu, X.; Liao, M.; Golberg, D. ZnO and ZnS Nanostructures: Ultraviolet-Light Emitters, Lasers, and Sensors. Crit. Rev. Solid State Mater. Sci. 2009, 34, 190–223. [Google Scholar] [CrossRef]
- Wang, X.; Huang, H.; Liang, B.; Liu, Z.; Chen, D.; Shen, G. ZnS Nanostructures: Synthesis, Properties, and Applications. Crit. Rev. Solid State Mater. Sci. 2013, 38, 57–90. [Google Scholar] [CrossRef]
- Kumar, S.; Fossard, F.; Amiri, G.; Chauveau, J.M.; Sallet, V. Induced structural modifications in ZnS nanowires via physical state of catalyst: Highlights of 15R crystal phase. Nano Res. 2022, 15, 377–385. [Google Scholar] [CrossRef]
- Jaquez, M.; Yu, K.M.; Ting, M.; Hettick, M.; Sánchez-Royo, J.F.; Wełna, M.; Javey, A.; Dubon, O.D.; Walukiewicz, W. Growth and characterization of ZnO1−xSx highly mismatched alloys over the entire composition. J. Appl. Phys. 2015, 118, 215702. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gao, P.; Li, J.; Summers, C.J.; Wang, Z. LRectangular Porous ZnO–ZnS Nanocables and ZnS Nanotubes. Adv. Mater. 2002, 14, 1732–1735. [Google Scholar] [CrossRef]
- Yan, C.; Xue, D. Room Temperature Fabrication of Hollow ZnS and ZnO Architectures by a Sacrificial Template Route. J. Phys. Chem. B 2006, 110, 7102–7106. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Qian, H.; Liu, Y.; Du, G.; Zhang, F.; Wang, L.; Hu, X. A microwave-assisted rapid route to synthesize ZnO/ZnS core-shell nanostructures via controllable surface sulfidation of ZnO nanorods. CrystEngComm 2011, 13, 3438–3443. [Google Scholar] [CrossRef]
- Jia, W.; Jia, B.; Qu, F.; Wu, X. Towards a highly efficient simulated sunlight driven photocatalyst: A case of heterostructured ZnO/ZnS hybrid structure. Dalton Trans. 2013, 42, 14178–14187. [Google Scholar] [CrossRef] [PubMed]
- Sadollahkhani, A.; Kazeminezhad, I.; Lu, J.; Nur, O.; Hultman, L.; Willander, M. Synthesis, structural characterization and photocatalytic application of ZnO@ZnS core-shell nanoparticles. RSC Adv. 2014, 4, 36940–36950. [Google Scholar] [CrossRef] [Green Version]
- Daiko, Y.; Schmidt, J.; Kawamura, G.; Romeis, S.; Segets, D.; Iwamoto, Y.; Peukert, W. Mechanochemically induced sulfur doping in ZnO via oxygen vacancy formation. Phys. Chem. Chem. Phys. 2017, 19, 13838–13845. [Google Scholar] [CrossRef]
- de Moraes, N.P.; Marins, L.G.P.; de Moura Yamanaka, M.Y.; Bacani, R.; da Silva Rocha, R.; Rodrigues, L.A. Efficient photodegradation of 4-chlorophenol under solar radiation using a new ZnO/ZnS/carbon xerogel composite as a photocatalyst. J. Photochem. Photobiol. A Chem. 2021, 418, 113377. [Google Scholar] [CrossRef]
- Giri, A.K.; Charan, C.; Saha, A.; Shahi, V.K.; Panda, A.B. An amperometric cholesterol biosensor with excellent sensitivity and limit of detection based on an enzyme-immobilized microtubular ZnO@ZnS heterostructure. J. Mater. Chem. A 2014, 2, 16997–17004. [Google Scholar] [CrossRef]
- Tarish, S.; Xu, Y.; Wang, Z.; Mate, F.; Al-Haddad, A.; Wang, W.; Lei, Y. Highly efficient biosensors by using well-ordered ZnO/ZnS core/shell nanotube arrays. Nanotechnology 2017, 28, 405501. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.-Y.; Song, J.; Lu, M.-P.; Lee, C.-Y.; Chen, L.-J.; Wang, Z.L. ZnO−ZnS Heterojunction and ZnS Nanowire Arrays for Electricity Generation. ACS Nano 2009, 3, 357–362. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, C.; Zhai, T.; Li, S.L.; Wang, X.; Liu, J.; Jie, X.; Liu, D.; Liao, M.; Koide, Y.; et al. Flexible Ultraviolet Photodetectors with Broad Photoresponse Based on Branched ZnS-ZnO Heterostructure Nanofilms. Adv. Mater. 2014, 26, 3088–3093. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.F.; Fan, D.H.; Shen, W.Z. A General Chemical Conversion Route To Synthesize Various ZnO-Based Core/Shell Structures. J. Phys. Chem. C 2008, 112, 10402–10406. [Google Scholar] [CrossRef]
- Chen, P.; Gu, L.; Cao, X. From single ZnO multipods to heterostructured ZnO/ZnS, ZnO/ZnSe, ZnO/Bi2S3 and ZnO/Cu2S multipods: Controlled synthesis and tunable optical and photoelectrochemical properties. CrystEngComm 2010, 12, 3950–3958. [Google Scholar] [CrossRef]
- Xitao, W.; Rong, L.; Kang, W. Synthesis of ZnO@ZnS-Bi2S3 core-shell nanorod grown on reduced graphene oxide sheets and its enhanced photocatalytic performance. J. Mater. Chem. A 2014, 2, 8304–8313. [Google Scholar] [CrossRef]
- Sundararajan, M.; Sakthivel, P.; Fernandez, A.C. Structural, optical and electrical properties of ZnO-ZnS nanocomposites prepared by simple hydrothermal method. J. Alloy. Compd. 2018, 768, 553–562. [Google Scholar] [CrossRef]
- Benyahia, K.; Djeffal, F.; Ferhati, H.; Benhaya, A.; Bendjerad, A.; Djaballah, Y.; Martin, N. Microstructured ZnO-ZnS composite for earth-abundant photovoltaics: Elaboration, surface analysis and enhanced optical performances. Sol. Energy 2021, 218, 312–319. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL14 User’s Manual; University of Torino: Torino, Italy, 2009. [Google Scholar]
- Dovesi, R.; Orlando, R.; Civalleri, B.; Roetti, C.; Saunders Victor, R.; Zicovich-Wilson Claudio, M. CRYSTAL: A computational tool for the ab initio study of the electronic properties of crystals. Z. Für Krist.-Cryst. Mater. 2005, 220, 571–573. [Google Scholar] [CrossRef]
- Doll, K.; Dovesi, R.; Orlando, R. Analytical Hartree–Fock gradients with respect to the cell parameter for systems periodic in three dimensions. Theor. Chem. Acc. 2004, 112, 394–402. [Google Scholar] [CrossRef] [Green Version]
- Zagorac, D.; Zagorac, J.; Schön, J.C.; Stojanovic, N.; Matovic, B. ZnO/ZnS (hetero)structures: Ab initio investigations of polytypic behavior of mixed ZnO and ZnS compounds. Acta Crystallogr. B 2018, 74, 628–642. [Google Scholar] [CrossRef]
- Zagorac, D.; Schön, J.C.; Zagorac, J.; Jansen, M. Prediction of structure candidates for zinc oxide as a function of pressure and investigation of their electronic properties. Phys. Rev. B 2014, 89, 075201. [Google Scholar] [CrossRef]
- Zagorac, D.; Schön, J.C.; Zagorac, J.; Jansen, M. Theoretical investigations of novel zinc oxide polytypes and in-depth study of their electronic properties. RSC Adv. 2015, 5, 25929–25935. [Google Scholar] [CrossRef] [Green Version]
- Schön, J.C.; Čančarević, Ž.; Jansen, M. Structure prediction of high-pressure phases for alkali metal sulfides. J. Chem. Phys. 2004, 121, 2289–2304. [Google Scholar] [CrossRef] [PubMed]
- Zagorac, D.; Doll, K.; Zagorac, J.; Jordanov, D.; Matović, B. Barium Sulfide under Pressure: Discovery of Metastable Polymorphs and Investigation of Electronic Properties on ab Initio Level. Inorg. Chem. 2017, 56, 10644–10654. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, D.; Zagorac, D.; Matovic, B.; Zarubica, A.; Zagorac, J. Anion substitution and influence of sulfur on the crystal structures, phase transitions, and electronic properties of mixed TiO2/TiS2 compounds. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2021, 77, 833–847. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Jaffe, J.E.; Hess, A.C. Hartree-Fock study of phase changes in ZnO at high pressure. Phys. Rev. B 1993, 48, 7903–7909. [Google Scholar] [CrossRef]
- Homann, T.; Hotje, U.; Binnewies, M.; Börger, A.; Becker, K.-D.; Bredow, T. Composition-dependent band gap in ZnSxSe1−x: A combined experimental and theoretical study. Solid State Sci. 2006, 8, 44–49. [Google Scholar] [CrossRef]
- Towler, M.D.; Allan, N.L.; Harrison, N.M.; Saunders, V.R.; Mackrodt, W.C.; Aprà, E. Ab initio study of MnO and NiO. Phys. Rev. B 1994, 50, 5041–5054. [Google Scholar] [CrossRef]
- Ferrari, A.M.; Pisani, C. An ab Initio Periodic Study of NiO Supported at the Pd(100) Surface. Part 1: The Perfect Epitaxial Monolayer. J. Phys. Chem. B 2006, 110, 7909–7917. [Google Scholar] [CrossRef] [PubMed]
- Mian, M.; Harrison, N.M.; Saunders, V.R.; Flavell, W.R. An ab initio Hartree-Fock investigation of galena (PbS). Chem. Phys. Lett. 1996, 257, 627–632. [Google Scholar] [CrossRef]
- Zagorac, D.; Doll, K.; Schön, J.C.; Jansen, M. Ab initio structure prediction for lead sulfide at standard and elevated pressures. Phys. Rev. B 2011, 84, 045206. [Google Scholar] [CrossRef]
- Hundt, R. KPLOT, A Program for Plotting and Analyzing Crystal Structures; Technicum Scientific Publishing: Stuttgart, Germany, 2016. [Google Scholar]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Turner, P. Xmgrace. Available online: https://plasma-gate.weizmann.ac.il/Grace/ (accessed on 5 May 2022).
- Chao, G.Y.; Gault, R.A. The occurrence of two rare polytypes of wurtzite, 4H and 8H, at Mont Saint-Hilaire, Quebec. Can. Mineral. 1998, 36, 775–778. [Google Scholar]
- Haussühl, S.; Müller, G. Neue ZnS-Polytypen (9R, 12R und 21R) in mesozoischen Sedimenten NW-Deutschlands. Beitraege Zur Mineral. Und Petrogr. 1963, 9, 28–39. [Google Scholar] [CrossRef]
- Skinner, B.J.; Barton, P.B. The substitution of oxygen for sulfur in wurtzite and sphalerite. Am. Mineral. 1960, 4, 612–615. [Google Scholar]
- Patil, A.B.; Patil, K.R.; Pardeshi, S.K. Ecofriendly synthesis and solar photocatalytic activity of S-doped ZnO. J. Hazard. Mater. 2010, 183, 315–323. [Google Scholar] [CrossRef]
- Chen, L.-C.; Tu, Y.-J.; Wang, Y.-S.; Kan, R.-S.; Huang, C.-M. Characterization and photoreactivity of N-, S-, and C-doped ZnO under UV and visible light illumination. J. Photochem. Photobiol. A Chem. 2008, 199, 170–178. [Google Scholar] [CrossRef]
- Shen, G.; Cho, J.H.; Yoo, J.K.; Yi, G.-C.; Lee, C.J. Synthesis and Optical Properties of S-Doped ZnO Nanostructures: Nanonails and Nanowires. J. Phys. Chem. B 2005, 109, 5491–5496. [Google Scholar] [CrossRef]
- Zafar, M.; Ahmed, S.; Shakil, M.; Choudhary, M.A. First-principles calculations of structural, electronic, and thermodynamic properties of ZnO1−xSx alloys. Chin. Phys. B 2014, 23, 106108. [Google Scholar] [CrossRef]
- Luo, K.; Sun, Y.; Zhou, L.; Wang, F.; Wu, F. Theoretical simulation of performances in CIGS thin-film solar cells with cadmium-free buffer layer. J. Semicond. 2017, 38, 084006. [Google Scholar] [CrossRef]
- Shabbir, S.; Shaari, A.; Ul Haq, B.; Ahmed, R.; AlFaify, S.; Ahmed, M.; Laref, A. First-principles investigations of structural parameters, electronic structures and optical spectra of 5–5- and BeO-type of ZnO1−xSx alloys. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 2020, 262, 114697. [Google Scholar] [CrossRef]
- Hou, Q.; Qi, M.; Li, Y. Effects of p-type conductive properties of triaxial strain-regulated ZnO (S, Se, Te) system. Phys. Scr. 2021, 96, 125815. [Google Scholar] [CrossRef]
- Srot, V.; Recnik, A.; Scheu, C.; Sturm, S.; Mirtic, B. Stacking faults and twin boundaries in sphalerite crystals from the Trepča mines in Kosovo. Am. Mineral. 2003, 88, 1809–1816. [Google Scholar] [CrossRef]
- Sokol, E.V.; Kokh, S.N.; Seryotkin, Y.V.; Deviatiiarova, A.S.; Goryainov, S.V.; Sharygin, V.V.; Khoury, H.N.; Karmanov, N.S.; Danilovsky, V.A.; Artemyev, D.A. Ultrahigh-Temperature Sphalerite from Zn-Cd-Se-Rich Combustion Metamorphic Marbles, Daba Complex, Central Jordan: Paragenesis, Chemistry, and Structure. Minerals 2020, 10, 822. [Google Scholar] [CrossRef]
- Huso, J.; Ritter, J.R.; Bergman, L.; McCluskey, M.D. High Order Oxygen Local Vibrational Modes in ZnS1−xOx. Phys. Status Solidi (b) 2019, 256, 1800607. [Google Scholar] [CrossRef]
- Müller, U. Inorganic Structural Chemistry; Wiley-VCH: Marburg, Germany, 2007. [Google Scholar]
- Boutaiba, F.; Belabbes, A.; Ferhat, M.; Bechstedt, F. Polytypism in ZnS, ZnSe, and ZnTe: First-principles study. Phys. Rev. B 2014, 89, 245308. [Google Scholar] [CrossRef] [Green Version]
- Mardix, S. Polytypism: A controlled thermodynamic phenomenon. Phys. Rev. B 1986, 33, 8677–8684. [Google Scholar] [CrossRef] [Green Version]
- Shabbir, S.; Shaari, A.; Ul Haq, B.; Ahmed, R.; AlFaify, S.; Ahmed, M.; Laref, A. First-principles investigations of electronic structures and optical spectra of wurtzite and sphalerite types of ZnO1−xSx (x = 0, 0.25, 0.50, 0.75 &1) alloys. Mater. Sci. Semicond. Process. 2021, 121, 105326. [Google Scholar] [CrossRef]
- Locmelis, S.; Brünig, C.; Binnewies, M.; Börger, A.; Becker, K.D.; Homann, T.; Bredow, T. Optical band gap in the system ZnO1−xSx. An experimental and quantum chemical study. J. Mater. Sci. 2007, 42, 1965–1971. [Google Scholar] [CrossRef]
- Baldissera, G.; Persson, C. Understanding the optical properties of ZnO1−xSx and ZnO1−xSex alloys. J. Appl. Phys. 2016, 119, 045704. [Google Scholar] [CrossRef] [Green Version]
- Stoliaroff, A.; Latouche, C. Accurate Ab Initio Calculations on Various PV-Based Materials: Which Functional to Be Used? J. Phys. Chem. C 2020, 124, 8467–8478. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, G.; Cao, Y.; Chen, J.; Shen, K.; Kumar, A.; Xu, M.; Li, Q.; Xu, Q. The magnetic and adsorption properties of ZnO1−xSx nanoparticles. Phys. Chem. Chem. Phys. 2017, 19, 26918–26925. [Google Scholar] [CrossRef]
- Meyer, B.K.; Polity, A.; Farangis, B.; He, Y.; Hasselkamp, D.; Krämer, T.; Wang, C. Structural properties and bandgap bowing of ZnO1−xSx thin films deposited by reactive sputtering. Appl. Phys. Lett. 2004, 85, 4929–4931. [Google Scholar] [CrossRef]
- Menad, A.; Benmalti, M.E.; Zaoui, A.; Ferhat, M. Impact of polytypism on the ground state properties of zinc oxide: A first-principles study. Results Phys. 2020, 18, 103316. [Google Scholar] [CrossRef]
- Apaolaza, A.; Richard, D.; Tejerina, M.R. Experimental and ab initio study of the structural and optical properties of ZnO coatings: Performance of the DFT+U approach. Process. Appl. Ceram. 2020, 14, 362–371. [Google Scholar] [CrossRef]
- Sponza, L.; Goniakowski, J.; Noguera, C. Structural, electronic, and spectral properties of six ZnO bulk polymorphs. Phys. Rev. B 2015, 91, 075126. [Google Scholar] [CrossRef]
- Sponza, L.; Goniakowski, J.; Noguera, C. Confinement effects in ultrathin ZnO polymorph films: Electronic and optical properties. Phys. Rev. B 2016, 93, 195435. [Google Scholar] [CrossRef] [Green Version]
- Shabbir, S.; Shaari, A.; Ul Haq, B.; Ahmed, R.; Ahmed, M. Investigations of novel polymorphs of ZnO for optoelectronic applications. Optik 2020, 206, 164285. [Google Scholar] [CrossRef]
- Ul Haq, B.; AlFaify, S.; Alshahrani, T.; Ahmed, R.; Tahir, S.A.; Amjed, N.; Laref, A. Exploring optoelectronic properties of ZnO monolayers originated from NaCl- and GeP-like polymorphs: A first-principles study. Results Phys. 2020, 19, 103367. [Google Scholar] [CrossRef]
- Ul Haq, B.; AlFaify, S.; Alshahrani, T.; Ahmed, R.; Butt, F.K.; Ur Rehman, S.; Tariq, Z. Devising square- and hexagonal-shaped monolayers of ZnO for nanoscale electronic and optoelectronic applications. Sol. Energy 2020, 211, 920–927. [Google Scholar] [CrossRef]
- Khomyak, V.; Shtepliuk, I.; Khranovskyy, V.; Yakimova, R. Band-gap engineering of ZnO1−xSx films grown by rf magnetron sputtering of ZnS target. Vacuum 2015, 121, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.J.; Barnard, A.S.; Zacharias, M. ZnO nanowires and nanobelts: Shape selection and thermodynamic modeling. Appl. Phys. Lett. 2007, 90, 143116. [Google Scholar] [CrossRef]
- Gayen, R.N.; Sarkar, K.; Hussain, S.; Bhar, R.; Pal, A.K. ZnO films prepared by modified sol-gel technique. Indian J. Pure Appl. Phys. (IJPAP) 2011, 47, 470–477. [Google Scholar]
- Kapitanova, O.O.; Baranov, A.N.; Panin, G.N. Structural and Optical Properties of ZnO1−xSx Nanoparticles. J. Nanoelectron. Optoelectron. 2012, 7, 633–636. [Google Scholar] [CrossRef] [Green Version]
- Polity, A.; Meyer, B.K.; Krämer, T.; Wang, C.; Haboeck, U.; Hoffmann, A. ZnO based ternary transparent conductors. Phys. Status Solidi (a) 2006, 203, 2867–2872. [Google Scholar] [CrossRef]
- Alqahtani, S.M.; Usman, M.; Ahmed, S.S. Unusual bandgap bowing in highly mismatched ZnOS alloys: Atomistic tight-binding band anti-crossing model. J. Appl. Phys. 2019, 125, 235704. [Google Scholar] [CrossRef]
- Huso, J.; Bergman, L.; McCluskey, M.D. Bandgap of cubic ZnS1−xOx from optical transmission spectroscopy. J. Appl. Phys. 2019, 125, 075704. [Google Scholar] [CrossRef]
- Torabi, A.; Staroverov, V.N. Band Gap Reduction in ZnO and ZnS by Creating Layered ZnO/ZnS Heterostructures. J. Phys. Chem. Lett. 2015, 6, 2075–2080. [Google Scholar] [CrossRef]
- Schrier, J.; Demchenko, D.O.; Lin, W.; Alivisatos, A.P. Optical Properties of ZnO/ZnS and ZnO/ZnTe Heterostructures for Photovoltaic Applications. Nano Lett. 2007, 7, 2377–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisi, E.H.; Elcombe, M.M. u parameters for the wurtzite structure of ZnS and ZnO using powder neutron diffraction. Acta Crystallogr. Sect. C 1989, 45, 1867–1870. [Google Scholar] [CrossRef]
- Desgreniers, S.; Beaulieu, L.; Lepage, I. Pressure-induced structural changes in ZnS. Phys. Rev. B 2000, 61, 8726–8733. [Google Scholar] [CrossRef]
- Mardix, S.; Kiflawi, I.; Kalman, Z.H. Double polytype regions in ZnS crystals. Acta Crystallogr. Sect. B 1969, 25, 1586–1589. [Google Scholar] [CrossRef]
- Evans, H.T., Jr.; McKnight, E.T. New wurtzite polytypes from Joplin, Missouri. Am. Mineral. 1959, 44, 1210–1218. [Google Scholar]
- Sowa, H.; Ahsbahs, H. High-pressure X-ray investigation of zincite ZnO single crystals using diamond anvils with an improved shape. J. Appl. Cryst. 2006, 39, 169–175. [Google Scholar] [CrossRef]
- Kim, S.-K.; Jeong, S.-Y.; Cho, C.-R. Structural reconstruction of hexagonal to cubic ZnO films on Pt/Ti/SiO2/Si substrate by annealing. Appl. Phys. Lett. 2003, 82, 562–564. [Google Scholar] [CrossRef]
- Mang, A.; Reimann, K.; Rübenacke, S. Band gaps, crystal-field splitting, spin-orbit coupling, and exciton binding energies in ZnO under hydrostatic pressure. Solid State Commun. 1995, 94, 251–254. [Google Scholar] [CrossRef]
- Huang, Z.; Lü, T.-Y.; Wang, H.-Q.; Zheng, J.-C. Thermoelectric properties of the 3C, 2H, 4H, and 6H polytypes of the wide-band-gap semiconductors SiC, GaN, and ZnO. AIP Adv. 2015, 5, 097204. [Google Scholar] [CrossRef] [Green Version]
- Vogel, D.; Krüger, P.; Pollmann, J. Ab initio electronic-structure calculations for II-VI semiconductors using self-interaction-corrected pseudopotentials. Phys. Rev. B. 1995, 52, R14316. [Google Scholar] [CrossRef]
- Ong, K.P.; Singh, D.J.; Wu, P. Analysis of the thermoelectric properties of n-type ZnO. Phys. Rev. B 2011, 83, 115110. [Google Scholar] [CrossRef]
- Tran, F.; Blaha, P. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 2009, 102, 226401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, M.; Noor, N.A.; Sabir, B.; Ali, S.; Sajjad, M.; Hussain, F.; Khan, N.U.; Amin, B.; Khenata, R. Ab-initio study of fundamental properties of ternary ZnO1−xSx alloys by using special quasi-random structures. Comput. Mater. Sci. 2014, 91, 285–291. [Google Scholar] [CrossRef]
- Meyer, B.K.; Polity, A.; Farangis, B.; He, Y.; Hasselkamp, D.; Kramer, T.; Wang, C.; Haboeck, U.; Hoffmann, A. On the composition dependence of ZnO1−xSx. Phys. Status Solidi (c) 2004, 1, 694–697. [Google Scholar] [CrossRef]
- Karazhanov, S.Z.; Ravindran, P.; Grossner, U.; Kjekshus, A.; Fjellvag, H.; Svensson, B.G. Electronic structure and band parameters for ZnX (X = O, S, Se, Te). J. Cryst. Growth 2006, 287, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Semiconductors Other Than Group IV Elements and III-V Compounds; Madelung, O. (Ed.) Data in Science and Technology; Springer: Berlin, Germany, 1992. [Google Scholar] [CrossRef]
- Springer Handbook of Electronic and Photonic Materials; Kasap, S.; Capper, P. (Eds.) Springer: Boston, MA, USA, 2006. [Google Scholar] [CrossRef]
- Strehlow, W.H.; Cook, E.L. Compilation of energy band gaps in elemental and binary compound semiconductors and insulators. J. Phys. Chem. Ref. Data 1973, 2, 163. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.K.; Ramprasad, R. Strain-assisted bandgap modulation in Zn based II-VI semiconductors. Appl. Phys. Lett. 2012, 100, 241903. [Google Scholar] [CrossRef]
- Zakharov, O.; Rubio, A.; Blase, X.; Cohen, M.L.; Louie, S.G. Quasiparticle band structures of six ii-vi compounds: ZnS, ZnSe, ZnTe, CdS, CdSe, and CdTe. Phys. Rev. B 1994, 50, 10780. [Google Scholar] [CrossRef]
- Fleszar, A.; Hanke, W. Electronic structure of IIB−VI semiconductors in the GW approximation. Phys. Rev. B 2005, 71, 045207. [Google Scholar] [CrossRef]
- Koller, D.; Tran, F.; Blaha, P. Merits and limits of the modified Becke-Johnson exchange potential. Phys. Rev. B 2011, 83, 195134. [Google Scholar] [CrossRef] [Green Version]
- Arora, A.; Manoharan, S.S. Structural and photo luminescent properties of uncapped nanocrystalline Cd1−xZnxS solid solutions. Opt. Mater. 2008, 31, 176–180. [Google Scholar] [CrossRef]
- Pan, H.L.; Yang, T.; Yao, B.; Deng, R.; Sui, R.Y.; Gao, L.L.; Shen, D.Z. Characterization and properties of ZnO1−xSx alloy films fabricated by radio-frequency magnetron sputtering. Appl. Surf. Sci. 2010, 256, 4621. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Composition | 4H Polytype (nm) | ||
|---|---|---|---|
| PBE0 | HSE06 | Experiment | |
| ZnO | P63mc (no. 186) a = 0.325, c = 1.050 | P63mc (no. 186) a = 0.325, c = 1.051 | n/a |
| ZnO0.75S0.25 | P3m1 (no. 156) a = 0.339, c = 1.110 | P3m1 (no. 156) a = 0.339, c = 1.110 | n/a |
| ZnO0.5S0.5 | P3m1 (no. 156) a = 0.356, c = 1.156 | P3m1 (no. 156) a = 0.356, c = 1.157 | n/a |
| ZnO0.25S0.75 | P3m1 (no. 156) a = 0.372, c = 1.209 | P3m1 (no. 156) a = 0.372, c = 1.210 | n/a |
| ZnS | P63mc (no. 186) a = 0.385, c = 1.257 | P63mc (no. 186) a = 0.386, c = 1.259 | P63mc (no. 186) a = 0.382, c = 1.252 * |
| Chemical Composition | 15R Polytype (nm) | ||
|---|---|---|---|
| PBE0 | HSE06 | Experiment | |
| ZnO | R3mH (no. 160) a = 0.325, c = 3.944 | R3mH (no. 160) a = 0.325, c = 3.946 | n/a |
| ZnO0.8S0.2 | R3mH (no. 160) a = 0.336, c = 4.107 | R3mH (no. 160) a = 0.336, c = 4.109 | n/a |
| ZnO0.5S0.5 | Cm (no. 8) a = 2.895, b = 0.356, c = 0.615, β = 94.04 | Cm (no. 8) a = 2.896, b = 0.356, c = 0.616, β = 94.04 | n/a |
| ZnO0.4S0.6 | R3mH (no. 160) a = 0.363, c = 4.422 | R3mH (no. 160) a = 0.363, c = 4.425 | n/a |
| ZnS | R3mH (no. 160) a = 0.385, c = 4.719 | R3mH (no. 160) a = 0.386, c = 4.740 | R3mH (no. 160) a = 0.382, c = 4.680 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zagorac, D.; Zagorac, J.; Pejić, M.; Matović, B.; Schön, J.C. Band Gap Engineering of Newly Discovered ZnO/ZnS Polytypic Nanomaterials. Nanomaterials 2022, 12, 1595. https://doi.org/10.3390/nano12091595
Zagorac D, Zagorac J, Pejić M, Matović B, Schön JC. Band Gap Engineering of Newly Discovered ZnO/ZnS Polytypic Nanomaterials. Nanomaterials. 2022; 12(9):1595. https://doi.org/10.3390/nano12091595
Chicago/Turabian StyleZagorac, Dejan, Jelena Zagorac, Milan Pejić, Branko Matović, and Johann Christian Schön. 2022. "Band Gap Engineering of Newly Discovered ZnO/ZnS Polytypic Nanomaterials" Nanomaterials 12, no. 9: 1595. https://doi.org/10.3390/nano12091595