
Recent Application of Core-Shell Nanostructured Catalysts for CO2 Thermocatalytic Conversion Processes
 , , and
, , and
Abstract
:1. Introduction
2. Approaches to the Synthesis of Core-Shell Catalysts for CO2 Utilization
3. CO2 Conversion Processes and Products
3.1. CO2 Hydrogenation Reactions
3.1.1. CO2 Methanation
Core-Shell Nanostructured Catalysts for CO2 Methanation
3.1.2. CO2 Hydrogenation to Methanol
Core-Shell Nanostructured Catalyst for CO2 Hydrogenation to Methanol
3.2. CO2-Reforming Reactions
3.2.1. CO2 Dry Reforming of Methane
Core-Shell Nanostructured Catalyst for CO2 Reforming of Methane
4. Challenges and Outlook
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Qureshi, S.; Mumtaz, M.; Chong, F.K.; Mukhtar, A.; Saqib, S.; Ullah, S.; Mubashir, M.; Khoo, K.S.; Show, P.L. A review on sensing and catalytic activity of nano-catalyst for synthesis of one-step ammonia and urea: Challenges and perspectives. Chemosphere 2021, 291, 132806. [Google Scholar] [CrossRef] [PubMed]
- Koohestanian, E.; Sadeghi, J.; Mohebbi-Kalhori, D.; Shahraki, F.; Samimi, A. A novel process for CO2 capture from the flue gases to produce urea and ammonia. Energy 2018, 144, 279–285. [Google Scholar] [CrossRef]
- Das, S.; Pérez-Ramírez, J.; Gong, J.; Dewangan, N.; Hidajat, K.; Gates, B.C.; Kawi, S. Core-shell structured catalysts for thermocatalytic, photocatalytic, and electrocatalytic conversion of CO2. Chem. Soc. Rev. 2020, 49, 2937–3004. [Google Scholar] [CrossRef]
- Watanabe, A.; Nakamura, T. Carbon Dynamics in Coral Reefs. In Blue Carbon in Shallow Coastal Ecosystems; Springer: Singapore, 2019; pp. 273–293. [Google Scholar] [CrossRef]
- Fan, W.K.; Tahir, M. Recent trends in developments of active metals and heterogenous materials for catalytic CO2 hydrogenation to renewable methane: A review. J. Environ. Chem. Eng. 2021, 9, 105460. [Google Scholar] [CrossRef]
- Yousaf, M.; Mahmood, A.; Elkamel, A.; Rizwan, M.; Zaman, M. Techno-economic analysis of integrated hydrogen and methanol production process by CO2 hydrogenation. Int. J. Greenh. Gas Control 2022, 115, 103615. [Google Scholar] [CrossRef]
- Ojelade, O.A.; Zaman, S.F. A review on CO2 hydrogenation to lower olefins: Understanding the structure-property relationships in heterogeneous catalytic systems. J. CO2 Util. 2021, 47, 101506. [Google Scholar] [CrossRef]
- Liang, J.; Wu, Q.; Huang, Y.; Cao, R. Reticular frameworks and their derived materials for CO2 conversion by thermos-catalysis. EnergyChem 2021, 3, 100064. [Google Scholar] [CrossRef]
- Hao, L.; Xia, Q.; Zhang, Q.; Masa, J.; Sun, Z. Improving the performance of metal-organic frameworks for thermo-catalytic CO2 conversion: Strategies and perspectives. Chin. J. Catal. 2021, 42, 1903–1920. [Google Scholar] [CrossRef]
- Anwar, M.; Fayyaz, A.; Sohail, N.; Khokhar, M.; Baqar, M.; Yasar, A.; Rasool, K.; Nazir, A.; Raja, M.; Rehan, M.; et al. CO2 utilization: Turning greenhouse gas into fuels and valuable products. J. Environ. Manag. 2020, 260, 110059. [Google Scholar] [CrossRef]
- Saravanan, A.; Kumar, P.S.; Vo, D.-V.N.; Jeevanantham, S.; Bhuvaneswari, V.; Narayanan, V.A.; Yaashikaa, P.; Swetha, S.; Reshma, B. A comprehensive review on different approaches for CO2 utilization and conversion pathways. Chem. Eng. Sci. 2021, 236, 116515. [Google Scholar] [CrossRef]
- Zhang, L.; Song, Y.; Shi, J.; Shen, Q.; Hu, D.; Gao, Q.; Chen, W.; Kow, K.-W.; Pang, C.; Sun, N.; et al. Frontiers of CO2 Capture and Utilization (CCU) towards Carbon Neutrality. Adv. Atmos. Sci. 2022, 39, 1252–1270. [Google Scholar] [CrossRef]
- Ding, M.; Liu, X.; Ma, P.; Yao, J. Porous materials for capture and catalytic conversion of CO2 at low concentration. Coord. Chem. Rev. 2022, 465, 214576. [Google Scholar] [CrossRef]
- Khalil, M.; Kadja, G.T.; Ilmi, M.M. Advanced nanomaterials for catalysis: Current progress in fine chemical synthesis, hydrocarbon processing, and renewable energy. J. Ind. Eng. Chem. 2020, 93, 78–100. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Su, W.; Xing, Y. Photocatalytic CO2 reduction over copper-based materials: A review. J. CO2 Util. 2022, 61, 102056. [Google Scholar] [CrossRef]
- Gawande, M.B.; Goswami, A.; Asefa, T.; Guo, H.; Biradar, A.V.; Peng, D.-L.; Zboril, R.; Varma, R.S. Core-shell nanoparticles: Synthesis and applications in catalysis and electrocatalysis. Chem. Soc. Rev. 2015, 44, 7540–7590. [Google Scholar] [CrossRef]
- Mitchell, S.; Qin, R.; Zheng, N.; Pérez-Ramírez, J. Nanoscale engineering of catalytic materials for sustainable technologies. Nat. Nanotechnol. 2020, 16, 129–139. [Google Scholar] [CrossRef]
- Wang, L.; Wang, F. Design Strategy, Synthesis, and Mechanism of Ni Catalysts for Methane Dry Reforming Reaction: Recent Advances and Future Perspectives. Energy Fuels 2022, 36, 5594–5621. [Google Scholar] [CrossRef]
- Mustafa, A.; Shuai, Y.; Lougou, B.G.; Wang, Z.; Razzaq, S.; Zhao, J.; Shan, J. Progress and perspective of electrochemical CO2 reduction on Pd-based nanomaterials. Chem. Eng. Sci. 2021, 245, 116869. [Google Scholar] [CrossRef]
- Xie, H.; Chen, S.; Ma, F.; Liang, J.; Miao, Z.; Wang, T.; Wang, H.-L.; Huang, Y.; Li, Q. Boosting Tunable Syngas Formation via Electrochemical CO2 Reduction on Cu/In2O3 Core/Shell Nanoparticles. ACS Appl. Mater. Interfaces 2018, 10, 36996–37004. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, W.; Ding, L.; Wu, Z.; Gao, F. Au Nanocrystals@Defective Amorphous MnO2 Nanosheets Core/Shell Nanostructure with Effective CO2 Adsorption and Activation toward CO2 Electroreduction to CO. ACS Sustain. Chem. Eng. 2021, 9, 5230–5239. [Google Scholar] [CrossRef]
- He, Y.; Li, Y.; Zhang, J.; Wang, S.; Huang, D.; Yang, G.; Yi, X.; Lin, H.; Han, X.; Hu, W.; et al. Low-temperature strategy toward Ni-NC@Ni core-shell nanostructure with Single-Ni sites for efficient CO2 electroreduction. Nano Energy 2020, 77, 105010. [Google Scholar] [CrossRef]
- Shang, L.; Lv, X.; Shen, H.; Shao, Z.; Zheng, G. Selective carbon dioxide electroreduction to ethylene and ethanol by core-shell copper/cuprous oxide. J. Colloid Interface Sci. 2019, 552, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, W.; Wan, H.; Li, C.; An, W.; Sheng, X.; Liang, X.; Wang, X.; Ren, Y.; Zheng, X.; et al. Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction. Chin. Chem. Lett. 2021, 33, 2259–2269. [Google Scholar] [CrossRef]
- Wanga, M.; Rena, X.; Yuana, G.; Niua, X.; Xua, Q.; Gaoa, W.; Zhua, S.; Wangab, Q. Selective electroreduction of CO2 to CO over co-electrodeposited dendritic core-shell indium-doped Cu@Cu2O catalyst. J. CO2 Util. 2019, 37, 204–212. [Google Scholar] [CrossRef]
- Wang, P.; Yang, H.; Xu, Y.; Huang, X.; Wang, J.; Zhong, M.; Cheng, T.; Shao, Q. Synergized Cu/Pb Core/Shell Electrocatalyst for High-Efficiency CO2 Reduction to C2+ Liquids. ACS Nano 2020, 15, 1039–1047. [Google Scholar] [CrossRef]
- Atsbha, T.A.; Yoon, T.; Seongho, P.; Lee, C.-J. A review on the catalytic conversion of CO2 using H2 for synthesis of CO, methanol, and hydrocarbons. J. CO2 Util. 2020, 44, 101413. [Google Scholar] [CrossRef]
- Lee, D.-E.; Moru, S.; Bhosale, R.; Jo, W.-K.; Tonda, S. Cu–Ni core-shell bimetallic cocatalyst decorated polymeric carbon nitride for highly efficient and selective methane production from photocatalytic CO2 reduction. Appl. Surf. Sci. 2022, 599, 153973. [Google Scholar] [CrossRef]
- Hong, D.; Lyu, L.-M.; Koga, K.; Shimoyama, Y.; Kon, Y. Plasmonic Ag@TiO2 Core-shell Nanoparticles for Enhanced CO2 Photoconversion to CH4. ACS Sustain. Chem. Eng. 2019, 7, 18955–18964. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, L.; Sheng, J.; Tang, F.; Zeng, K.; Wang, L.; Liang, K.; Hu, H.; Liu, Y.-N. Photostable core-shell CdS/ZIF-8 composite for enhanced photocatalytic reduction of CO2. Appl. Surf. Sci. 2019, 498, 143899. [Google Scholar] [CrossRef]
- Pougin, A.; Dodekatos, G.; Dilla, M.; Tüysüz, H.; Strunk, J. Au@TiO2 Core-Shell Composites for the Photocatalytic Reduction of CO2. Chemistry 2018, 24, 12416–12425. [Google Scholar] [CrossRef]
- Kong, Z.-C.; Liao, J.-F.; Dong, Y.-J.; Xu, Y.-F.; Chen, H.-Y.; Kuang, D.-B.; Su, C.-Y. Core@Shell CsPbBr3@Zeolitic Imidazolate Framework Nanocomposite for Efficient Photocatalytic CO2 Reduction. ACS Energy Lett. 2018, 3, 2656–2662. [Google Scholar] [CrossRef]
- Tran, D.T.; Kshetri, T.; Chuong, N.D.; Gautam, J.; Van Hien, H.; Tuan, L.H.; Kim, N.H.; Le, H.T. Emerging core-shell nanostructured catalysts of transition metal encapsulated by two-dimensional carbon materials for electrochemical applications. Nano Today 2018, 22, 100–131. [Google Scholar] [CrossRef]
- Ateka, A.; Rodriguez-Vega, P.; Ereña, J.; Aguayo, A.; Bilbao, J. A review on the valorization of CO2. Focusing on the thermodynamics and catalyst design studies of the direct synthesis of dimethyl ether. Fuel Process. Technol. 2022, 233, 107310. [Google Scholar] [CrossRef]
- Ye, R.; Wang, X.; Price, C.; Liu, X.; Yang, Q.; Jaroniec, M.; Liu, J. Engineering of Yolk/Core-shell Structured Nanoreactors for Thermal Hydrogenations. Small 2020, 17, e1906250. [Google Scholar] [CrossRef]
- Shamzhy, M.; Opanasenko, M.; Concepción, P.; Martínez, A. New trends in tailoring active sites in zeolite-based catalysts. Chem. Soc. Rev. 2019, 48, 1095–1149. [Google Scholar] [CrossRef]
- Bo, Y.; Gao, C.; Xiong, Y. Recent advances in engineering active sites for photocatalytic CO2 reduction. Nanoscale 2020, 12, 12196–12209. [Google Scholar] [CrossRef]
- Etim, U.J.; Zhang, C.; Zhong, Z. Impacts of the Catalyst Structures on CO2 Activation on Catalyst Surfaces. Nanomaterials 2021, 11, 3265. [Google Scholar] [CrossRef]
- Gao, X.; Wang, Z.; Huang, Q.; Jiang, M.; Askari, S.; Dewangan, N.; Kawi, S. State-of-art modifications of heterogeneous catalysts for CO2 methanation—Active sites, surface basicity and oxygen defects. Catal. Today 2022, 402, 88–103. [Google Scholar] [CrossRef]
- Lee, J.D.; Miller, J.B.; Shneidman, A.V.; Sun, L.; Weaver, J.F.; Aizenberg, J.; Biener, J.; Boscoboinik, J.A.; Foucher, A.C.; Frenkel, A.I.; et al. Dilute Alloys Based on Au, Ag, or Cu for Efficient Catalysis: From Synthesis to Active Sites. Chem. Rev. 2022, 122, 8758–8808. [Google Scholar] [CrossRef] [PubMed]
- Vogt, C.; Weckhuysen, B.M. The concept of active site in heterogeneous catalysis. Nat. Rev. Chem. 2022, 6, 89–111. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Z.; Kawi, S. Sintering and Coke Resistant Core/Yolk Shell Catalyst for Hydrocarbon Reforming. ChemCatChem 2018, 11, 202–224. [Google Scholar] [CrossRef]
- Gao, X.; Wang, Z.; Ashok, J.; Kawi, S. A comprehensive review of anti-coking, anti-poisoning and anti-sintering catalysts for biomass tar reforming reaction. Chem. Eng. Sci. X 2020, 7, 100065. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef] [Green Version]
- Price, C.A.H.; Pastor-Perez, L.; Reina, T.R.; Liu, J. Yolk-Shell structured NiCo@SiO2 nanoreactor for CO2 upgrading via reverse water-gas shift reaction. Catal. Today 2020, 383, 358–367. [Google Scholar] [CrossRef]
- Murthy, P.S.; Liang, W.; Jiang, Y.; Huang, J. Cu-Based Nanocatalysts for CO2 Hydrogenation to Methanol. Energy Fuels 2021, 35, 8558–8584. [Google Scholar] [CrossRef]
- Ghosh Chaudhuri, R.; Paria, S. Core/Shell Nanoparticles: Classes, Properties, Synthesis Mechanisms, Characterization, and Applications. Chem. Rev. 2012, 112, 2373–2433. [Google Scholar] [CrossRef]
- Masoumifard, N.; Guillet-Nicolas, R.; Kleitz, F. Synthesis of Engineered Zeolitic Materials: From Classical Zeolites to Hierarchical Core-Shell Materials. Adv. Mater. 2018, 30, e1704439. [Google Scholar] [CrossRef]
- Zuo, B.; Li, W.; Wu, X.; Wang, S.; Deng, Q.; Huang, M. Recent Advances in the Synthesis, Surface Modifications and Applications of Core-Shell Magnetic Mesoporous Silica Nanospheres. Chem.-Asian J. 2020, 15, 1248–1265. [Google Scholar] [CrossRef]
- Galogahi, F.M.; Zhu, Y.; An, H.; Nguyen, N.-T. Core-shell microparticles: Generation approaches and applications. J. Sci. Adv. Mater. Devices 2020, 5, 417–435. [Google Scholar] [CrossRef]
- Yang, G.; Xing, C.; Hirohama, W.; Jin, Y.; Zeng, C.; Suehiro, Y.; Wang, T.; Yoneyama, Y.; Tsubaki, N. Tandem catalytic synthesis of light isoparaffin from syngas via Fischer–Tropsch synthesis by newly developed core–shell-like zeolite capsule catalysts. Catal. Today 2013, 215, 29–35. [Google Scholar] [CrossRef]
- Wang, Z.; Qi, J.; Yang, N.; Yu, R.; Wang, D. Core-shell nano/microstructures for heterogeneous tandem catalysis. Mater. Chem. Front. 2020, 5, 1126–1139. [Google Scholar] [CrossRef]
- Ma, Z.; Porosoff, M.D. Development of Tandem Catalysts for CO2 Hydrogenation to Olefins. ACS Catal. 2019, 9, 2639–2656. [Google Scholar] [CrossRef]
- Yin, X.; Yang, L.; Gao, Q. Core-shell nanostructured electrocatalysts for water splitting. Nanoscale 2020, 12, 15944–15969. [Google Scholar] [CrossRef]
- Grams, J.; Ruppert, A. Catalyst Stability—Bottleneck of Efficient Catalytic Pyrolysis. Catalysts 2021, 11, 265. [Google Scholar] [CrossRef]
- Kosinov, N.; Liu, C.; Hensen, E.J.M.; Pidko, E.A. Engineering of Transition Metal Catalysts Confined in Zeolites. Chem. Mater. 2018, 30, 3177–3198. [Google Scholar] [CrossRef]
- Shi, Z.; Tan, Q.; Wu, D. A novel Core-shell structured CuIn@SiO 2 catalyst for CO 2 hydrogenation to methanol. AIChE J. 2018, 65, 1047–1058. [Google Scholar] [CrossRef]
- Golunski, S.; Burch, R. CO2 Hydrogenation to Methanol over Copper Catalysts: Learning from Syngas Conversion. Top. Catal. 2021, 64, 974–983. [Google Scholar] [CrossRef]
- Navarro-Jaén, S.; Virginie, M.; Bonin, J.; Robert, M.; Wojcieszak, R.; Khodakov, A.Y. Highlights and challenges in the selective reduction of carbon dioxide to methanol. Nat. Rev. Chem. 2021, 5, 564–579. [Google Scholar] [CrossRef]
- Ilsemann, J.; Straß-Eifert, A.; Friedland, J.; Kiewidt, L.; Thöming, J.; Bäumer, M.; Güttel, R. Cobalt@Silica Core-Shell Catalysts for Hydrogenation of CO/CO 2 Mixtures to Methane. ChemCatChem 2019, 11, 4884–4893. [Google Scholar] [CrossRef]
- Xu, W.-L.; Liu, B.; Wang, Y.-C.; Xiao, G.-Y.; Chen, X.; Xu, W.-H.; Lu, Y.-P. A facile strategy for one-step hydrothermal preparation of porous hydroxyapatite microspheres with core-shell structure. J. Mater. Res. Technol. 2022, 17, 320–328. [Google Scholar] [CrossRef]
- Soltani, S.; Khanian, N.; Choong, T.S.Y.; Asim, N.; Zhao, Y. Microwave-assisted hydrothermal synthesis of sulfonated TiO2-GO core-shell solid spheres as heterogeneous esterification mesoporous catalyst for biodiesel production. Energy Convers. Manag. 2021, 238, 114165. [Google Scholar] [CrossRef]
- Fu, H.; Yang, X.; Wu, Z.; He, P.; Xiong, S.; Han, D.; An, X. Gas-sensing performance of In2O3@MoO3 hollow core-shell nanospheres prepared by a two-step hydrothermal method. Sens. Actuators B: Chem. 2021, 352, 131007. [Google Scholar] [CrossRef]
- Ye, H.; Wang, J.; Wang, W.-H.; Bao, M. Encapsulation of Pd sub-nanoclusters in SiO2 nanospheres by a reverse microemulsion method. Funct. Mater. Lett. 2018, 11, 1850081. [Google Scholar] [CrossRef]
- Al-Kinani, M.A.; Haider, A.J.; Al-Musawi, S. High Uniformity Distribution of Fe@Au Preparation by a Micro-Emulsion Method. IOP Conf. Ser. Mater. Sci. Eng. 2020, 987, 012013. [Google Scholar] [CrossRef]
- Wu, H.; Gao, C.; Ma, L.; Zhong, Y.; Chai, S.; Lu, S.; Liu, J.; Wang, L. Controllable microemulsion method for the synthesis of Mg(OH) 2 /PS core-shell structures. Micro Nano Lett. 2021, 16, 413–418. [Google Scholar] [CrossRef]
- Tojo, C.; Buceta, D.; López-Quintela, M.A. On the minimum reactant concentration required to prepare Au/M core-shell nanoparticles by the one-pot microemulsion route. Phys. Sci. Rev. 2019, 5, 15–20. [Google Scholar] [CrossRef]
- Xia, M.; Yao, Q.; Yang, H.; Guo, T.; Du, X.; Zhang, Y.; Li, G.; Luo, Y. Preparation of Bi2O3/Al Core-Shell Energetic Composite by Two-Step Ball Milling Method and Its Application in Solid Propellant. Materials 2019, 12, 1879. [Google Scholar] [CrossRef] [Green Version]
- Manoharan, K.; Sundaram, R.; Raman, K. Enhanced hydrogen storage and superior capacitive performances of ball milled PMMA/h-BN core-shell nanocomposite. Ionics 2022, 28, 2875–2894. [Google Scholar] [CrossRef]
- Chen, C.; Tuo, Y.; Lu, Q.; Lu, H.; Zhang, S.; Zhou, Y.; Zhang, J.; Liu, Z.; Kang, Z.; Feng, X.; et al. Hierarchical trimetallic Co-Ni-Fe oxides derived from core-shell structured metal-organic frameworks for highly efficient oxygen evolution reaction. Appl. Catal. B Environ. 2021, 287, 119953. [Google Scholar] [CrossRef]
- Qu, F.; Zhang, N.; Zhang, S.; Zhao, R.; Yao, D.; Ruan, S.; Yang, M. Construction of Co3O4/CoWO4 core-shell urchin-like microspheres through ion-exchange method for high-performance acetone gas sensing performance. Sen. Actuators B: Chem. 2020, 309, 127711. [Google Scholar] [CrossRef]
- Hu, J.; Galvita, V.V.; Poelman, H.; Marin, G.B. Advanced Chemical Looping Materials for CO2 Utilization: A Review. Materials 2018, 11, 1187. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, M.; Balaraman, E. Confinement of Nanoparticles in Carbon Nanotubes: A New Paradigm in Heterogeneous Catalysis. In Advanced Heterogeneous Catalysts Volume 1: Applications at the Nano-Scale; American Chemical Society: Washington, DC, USA, 2020; pp. 459–481. [Google Scholar] [CrossRef]
- Hongmanorom, P.; Ashok, J.; Chirawatkul, P.; Kawi, S. Interfacial synergistic catalysis over Ni nanoparticles encapsulated in mesoporous ceria for CO2 methanation. Appl. Catal. B Environ. 2021, 297, 120454. [Google Scholar] [CrossRef]
- Rui, N.; Zhang, X.; Zhang, F.; Liu, Z.; Cao, X.; Xie, Z.; Zou, R.; Senanayake, S.D.; Yang, Y.; Rodriguez, J.A.; et al. Highly active Ni/CeO2 catalyst for CO2 methanation: Preparation and characterization. Appl. Catal. B Environ. 2020, 282, 119581. [Google Scholar] [CrossRef]
- Price, C.A.H.; Reina, T.R.; Liu, J. Engineering heterogenous catalysts for chemical CO2 utilization: Lessons from thermal catalysis and advantages of yolk@shell structured nanoreactors. J. Energy Chem. 2020, 57, 304–324. [Google Scholar] [CrossRef]
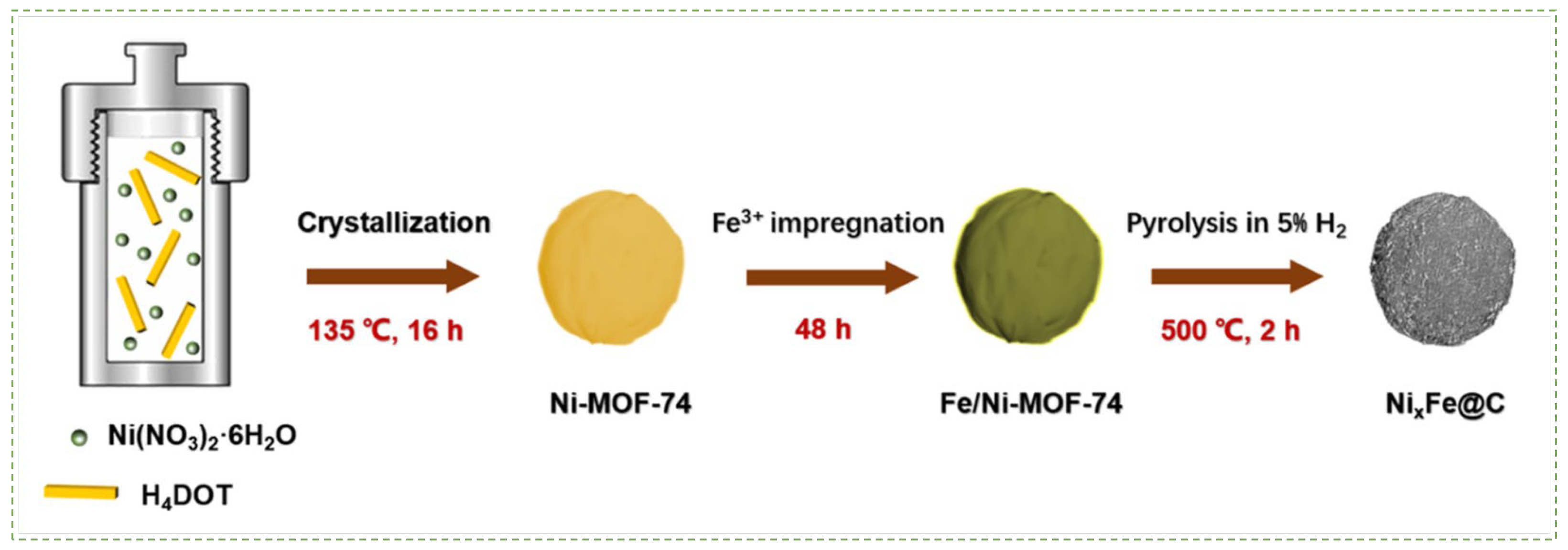
- Li, Y.-T.; Zhou, L.; Cui, W.-G.; Li, Z.-F.; Li, W.; Hu, T.-L. Iron promoted MOF-derived carbon encapsulated NiFe alloy nanoparticles core-shell catalyst for CO2 methanation. J. CO2 Util. 2022, 62, 102093. [Google Scholar] [CrossRef]
- Cui, W.-G.; Zhuang, X.-Y.; Li, Y.-T.; Zhang, H.; Dai, J.-J.; Zhou, L.; Hu, Z.; Hu, T.-L. Engineering Co/MnO heterointerface inside porous graphitic carbon for boosting the low-temperature CO2methanation. Appl. Catal. B Environ. 2021, 287, 119959. [Google Scholar] [CrossRef]
- Lee, W.J.; Li, C.; Prajitno, H.; Yoo, J.; Patel, J.; Yang, Y.; Lim, S. Recent trend in thermal catalytic low temperature CO2 methanation: A critical review. Catal. Today 2020, 368, 2–19. [Google Scholar] [CrossRef]
- Marinho, A.L.; Toniolo, F.S.; Noronha, F.B.; Epron, F.; Duprez, D.; Bion, N. Highly active and stable Ni dispersed on mesoporous CeO2-Al2O3 catalysts for production of syngas by dry reforming of methane. Appl. Catal. B Environ. 2020, 281, 119459. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, Y.; Liu, Q. Highly Efficient Ni-Phyllosilicate Catalyst with Surface and Interface Confinement for CO2 and CO Methanation. Ind. Eng. Chem. Res. 2021, 60, 6981–6992. [Google Scholar] [CrossRef]
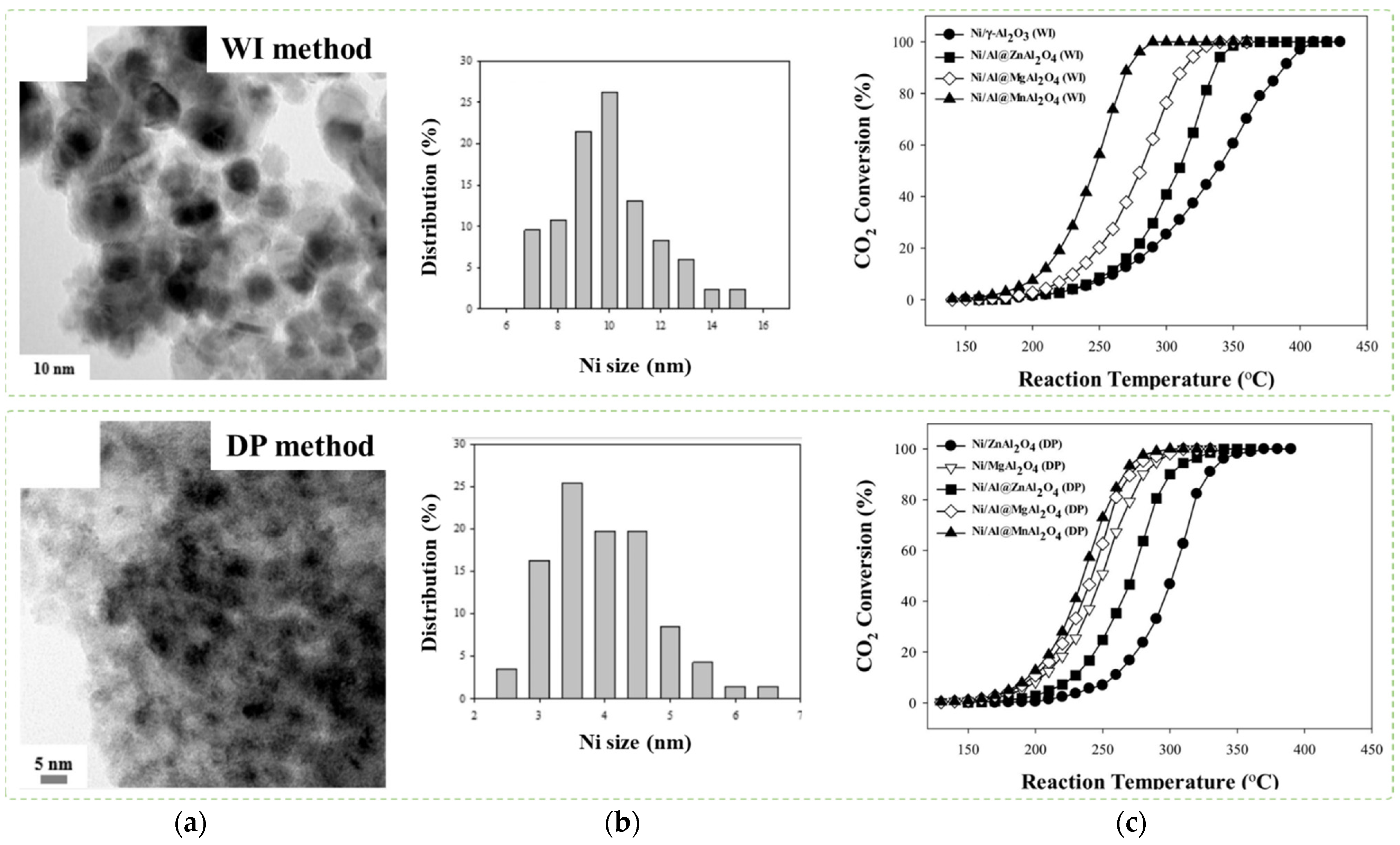
- Le, T.A.; Kim, J.; Kang, J.K.; Park, E.D. CO and CO methanation over Ni/Al@Al O3 core-shell catalyst. Catal. Today 2019, 356, 622–630. [Google Scholar] [CrossRef]
- Goodarzi, F.; Kang, L.; Wang, F.R.; Joensen, F.; Kegnaes, S.; Mielby, J. Methanation of Carbon Dioxide over Zeolite-Encapsulated Nickel Nanoparticles. ChemCatChem 2018, 10, 1566–1570. [Google Scholar] [CrossRef] [Green Version]
- Le, T.A.; Kim, J.; Jeong, Y.R.; Park, E.D. CO2 Methanation over Ni/Al@MAl2O4 (M = Zn, Mg, or Mn) Catalysts. Catalysts 2019, 9, 599. [Google Scholar] [CrossRef] [Green Version]
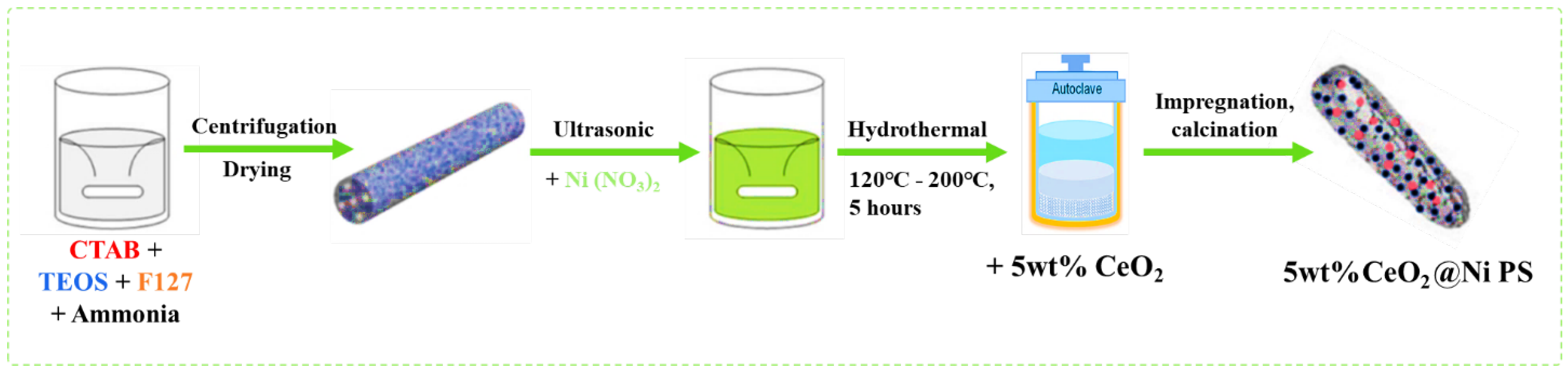
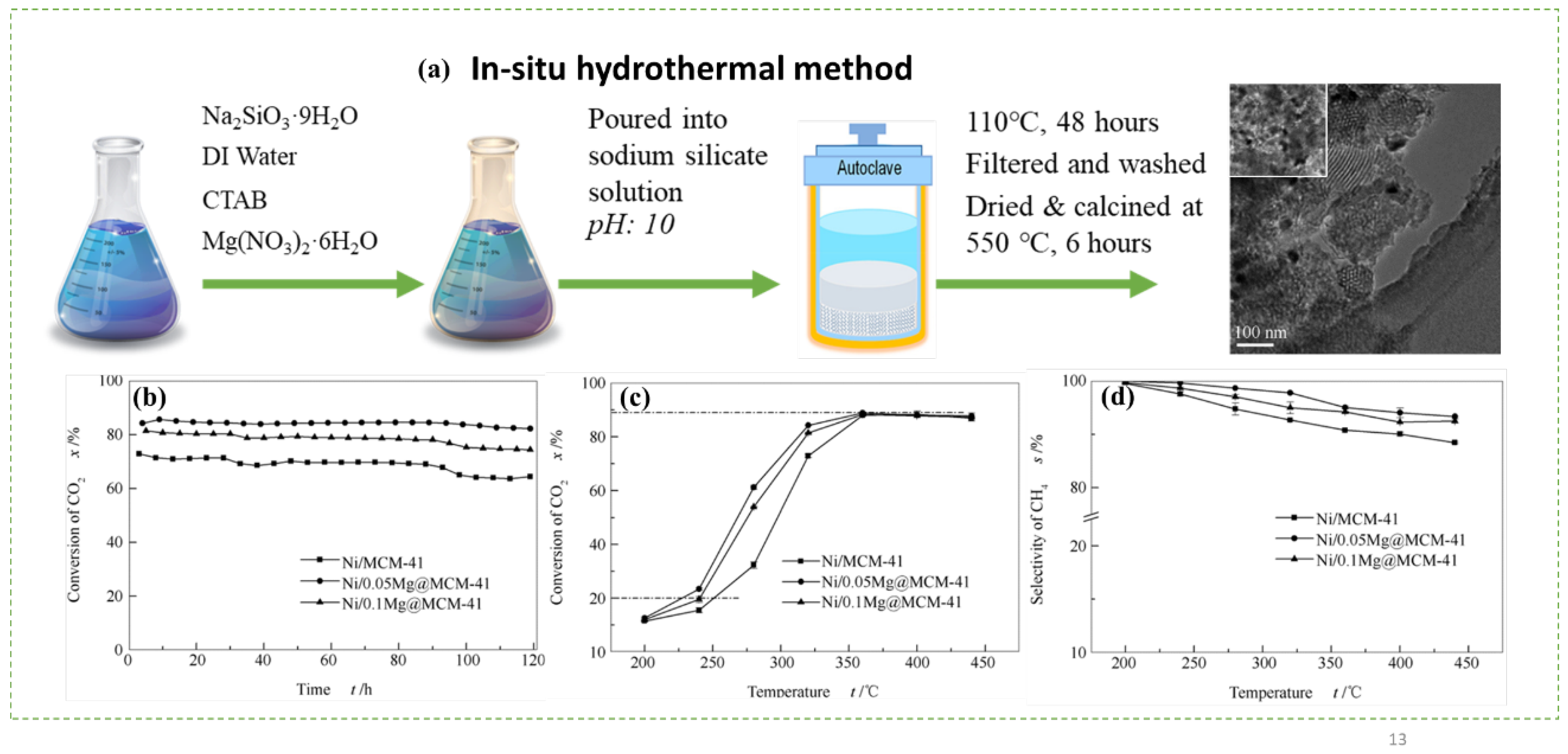
- Wang, X.-L.; Yang, M.; Zhu, L.-J.; Zhu, X.-N.; Wang, S.-R. CO2 methanation over Ni/Mg@MCM-41 prepared by in-situ synthesis method. J. Fuel Chem. Technol. 2020, 48, 456–465. [Google Scholar] [CrossRef]
- Han, Y.; Wen, B.; Zhu, M. Core-Shell Structured Ni@SiO2 Catalysts Exhibiting Excellent Catalytic Performance for Syngas Methanation Reactions. Catalysts 2017, 7, 21. [Google Scholar] [CrossRef]
- Shi, C.; Elgarni, M.; Mahinpey, N. Process design and simulation study: CO2 utilization through mixed reforming of methane for methanol synthesis. Chem. Eng. Sci. 2020, 233, 116364. [Google Scholar] [CrossRef]
- Battaglia, P.; Buffo, G.; Ferrero, D.; Santarelli, M.; Lanzini, A. Methanol synthesis through CO2 capture and hydrogenation: Thermal integration, energy performance and techno-economic assessment. J. CO2 Util. 2020, 44, 101407. [Google Scholar] [CrossRef]
- Nielsen, N.D.; Jensen, A.D.; Christensen, J.M. The roles of CO and CO2 in high pressure methanol synthesis over Cu-based catalysts. J. Catal. 2020, 393, 324–334. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.-X.; Zijlstra, B.; Filot, I.A.; Zhou, Z.; Sun, S.; Hensen, E.J. Optimum Cu nanoparticle catalysts for CO2 hydrogenation towards methanol. Nano Energy 2018, 43, 200–209. [Google Scholar] [CrossRef]
- Mitsuka, Y.; Ogiwara, N.; Mukoyoshi, M.; Kitagawa, H.; Yamamoto, T.; Toriyama, T.; Matsumura, S.; Haneda, M.; Kawaguchi, S.; Kubota, Y.; et al. Fabrication of Integrated Copper-Based Nanoparticles/Amorphous Metal–Organic Framework by a Facile Spray-Drying Method: Highly Enhanced CO 2 Hydrogenation Activity for Methanol Synthesis. Angew. Chem. Int. Ed. 2021, 60, 22283–22288. [Google Scholar] [CrossRef]
- Cui, X.; Chen, S.; Yang, H.; Liu, Y.; Wang, H.; Zhang, H.; Xue, Y.; Wang, G.; Niu, Y.; Deng, T.; et al. Improving methanol selectivity in CO2 hydrogenation by tuning the distance of Cu on catalyst. Appl. Catal. B Environ. 2021, 298, 120590. [Google Scholar] [CrossRef]
- Cui, X.; Van Muyden, A.P.; Dyson, P.J. Utility of Core-shell Nanomaterials in the Catalytic Transformations of Renewable Substrates. Chem.-A Eur. J. 2020, 27, 12–19. [Google Scholar] [CrossRef]
- Jangam, A.; Hongmanorom, P.; Wai, M.H.; Poerjoto, A.J.; Xi, S.; Borgna, A.; Kawi, S. CO2 Hydrogenation to Methanol over Partially Reduced Cu-SiO2P Catalysts: The Crucial Role of Hydroxyls for Methanol Selectivity. ACS Appl. Energy Mater. 2021, 4, 12149–12162. [Google Scholar] [CrossRef]
- Onishi, N.; Himeda, Y. CO2 hydrogenation to methanol by organometallic catalysts. Chem Catal. 2022, 2, 242–252. [Google Scholar] [CrossRef]
- An, B.; Zhang, J.; Cheng, K.; Ji, P.; Wang, C.; Lin, W. Confinement of Ultrasmall Cu/ZnOx Nanoparticles in Metal–Organic Frameworks for Selective Methanol Synthesis from Catalytic Hydrogenation of CO2. J. Am. Chem. Soc. 2017, 139, 3834–3840. [Google Scholar] [CrossRef]
- Tisseraud, C.; Comminges, C.; Belin, T.; Ahouari, H.; Soualah, A.; Pouilloux, Y.; Le Valant, A. The Cu-ZnO synergy in methanol synthesis from CO2, Part 2: Origin of the methanol and CO selectivities explained by experimental studies and a sphere contact quantification model in randomly packed binary mixtures on Cu–ZnO coprecipitate catalysts. J. Catal. 2015, 330, 533–544. [Google Scholar] [CrossRef]
- Tisseraud, C.; Comminges, C.; Habrioux, A.; Pronier, S.; Pouilloux, Y.; Le Valant, A. Cu-ZnO catalysts for CO2 hydrogenation to methanol: Morphology change induced by ZnO lixiviation and its impact on the active phase formation. Mol. Catal. 2018, 446, 98–105. [Google Scholar] [CrossRef]
- Le Valant, A.; Comminges, C.; Tisseraud, C.; Canaff, C.; Pinard, L.; Pouilloux, Y. The Cu-ZnO synergy in methanol synthesis from CO2, Part 1: Origin of active site explained by experimental studies and a sphere contact quantification model on Cu + ZnO mechanical mixtures. J. Catal. 2015, 324, 41–49. [Google Scholar] [CrossRef]
- Yang, H.; Gao, P.; Zhang, C.; Zhong, L.; Li, X.; Wang, S.; Wang, H.; Wei, W.; Sun, Y. Core-shell structured Cu@m-SiO2 and Cu/ZnO@m-SiO2 catalysts for methanol synthesis from CO2 hydrogenation. Catal. Commun. 2016, 84, 56–60. [Google Scholar] [CrossRef]
- Han, X.; Li, M.; Chang, X.; Hao, Z.; Chen, J.; Pan, Y.; Kawi, S.; Ma, X. Hollow structured Cu@ZrO2 derived from Zr-MOF for selective hydrogenation of CO2 to methanol. J. Energy Chem. 2022, 71, 277–287. [Google Scholar] [CrossRef]
- Xiao, X.; Gao, J.; Xi, S.; Lim, S.H.; Png, A.K.W.; Borgna, A.; Chu, W.; Liu, Y. Experimental and in situ DRIFTs studies on confined metallic copper stabilized Pd species for enhanced CO2 reduction to formate. Appl. Catal. B Environ. 2022, 309, 121239. [Google Scholar] [CrossRef]
- Kosari, M.; Anjum, U.; Xi, S.; Lim, A.M.H.; Seayad, A.M.; Raj, E.A.J.; Kozlov, S.M.; Borgna, A.; Zeng, H.C. Revamping SiO2 Spheres by Core-shell Porosity Endowment to Construct a Mazelike Nanoreactor for Enhanced Catalysis in CO 2 Hydrogenation to Methanol. Adv. Funct. Mater. 2021, 31, 2102896. [Google Scholar] [CrossRef]
- Gao, X.; Ge, Z.; Zhu, G.; Wang, Z.; Ashok, J.; Kawi, S. Anti-Coking and Anti-Sintering Ni/Al2O3 Catalysts in the Dry Reforming of Methane: Recent Progress and Prospects. Catalysts 2021, 11, 1003. [Google Scholar] [CrossRef]
- Pan, C.; Guo, Z.; Dai, H.; Ren, R.; Chu, W. Anti-sintering mesoporous Ni–Pd bimetallic catalysts for hydrogen production via dry reforming of methane. Int. J. Hydrogen Energy 2020, 45, 16133–16143. [Google Scholar] [CrossRef]
- Sharifianjazi, F.; Esmaeilkhanianb, A.; Bazlic, L.; Eskandarinezhadd, S.; Parisa, S.; Mohammad, Y.; Bawadi, A.; Peyman, A.; Salahshoura, P.; Sadeghih, F. A review on recent advances in dry reforming of methane over Ni- and Co-based nanocatalysts. Int. J. Hydrogen Energy 2021. [Google Scholar] [CrossRef]
- Kim, S.; Lauterbach, J.; Sasmaz, E. Yolk–Shell Pt-NiCe@SiO2 Single-Atom-Alloy Catalysts for Low-Temperature Dry Reforming of Methane. ACS Catal. 2021, 11, 8247–8260. [Google Scholar] [CrossRef]
- Chen, H.; Chansai, S.; Xu, S.; Xu, S.; Mu, Y.; Hardacre, C.; Fan, X. Dry reforming of methane on bimetallic Pt–Ni@CeO2 catalyst: A in situ DRIFTS-MS mechanistic study. Catal. Sci. Technol. 2021, 11, 5260–5272. [Google Scholar] [CrossRef]
- Shen, D.; Li, Z.; Shan, J.; Yu, G.; Wang, X.; Zhang, Y.; Liu, C.; Lyu, S.; Li, J.; Li, L. Synergistic Pt-CeO2 interface boosting low temperature dry reforming of methane. Appl. Catal. B Environ. 2022, 318, 121809. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, Y.; Xie, B.; Xia, S. Theoretical investigation onto the reaction mechanism of dry reforming of methane on core-shell Cu-Ni-Pt ternary alloy clusters. Chem. Phys. Lett. 2021, 781, 138975. [Google Scholar] [CrossRef]
- Han, K.; Wang, S.; Liu, Q.; Wang, F. Optimizing the Ni/Cu Ratio in Ni–Cu Nanoparticle Catalysts for Methane Dry Reforming. ACS Appl. Nano Mater. 2021, 4, 5340–5348. [Google Scholar] [CrossRef]
- Marinho, A.L.; Rabelo-Neto, R.C.; Epron, F.; Bion, N.; Toniolo, F.S.; Noronha, F.B. Embedded Ni nanoparticles in CeZrO2 as stable catalyst for dry reforming of methane. Appl. Catal. B Environ. 2020, 268, 118387. [Google Scholar] [CrossRef]
- Han, B.; Zhao, L.; Wang, F.; Xu, L.; Yu, H.; Cui, Y.; Zhang, J.; Shi, W. Effect of Calcination Temperature on the Performance of the Ni@SiO2 Catalyst in Methane Dry Reforming. Ind. Eng. Chem. Res. 2020, 59, 13370–13379. [Google Scholar] [CrossRef]
- Chong, C.C.; Cheng, Y.W.; Bahari, M.B.; Teh, L.P.; Abidin, S.Z.; Setiabudi, H.D. Development of nanosilica-based catalyst for syngas production via CO2 reforming of CH4: A review. Int. J. Hydrogen Energy 2020, 46, 24687–24708. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Y.; Gao, Z.; Zhang, X.; Zhang, L.; Wang, M.; Chen, B.; Diao, Y.; Li, Y.; Xiao, D.; et al. Embedding high loading and uniform Ni nanoparticles into silicalite-1 zeolite for dry reforming of methane. Appl. Catal. B Environ. 2022, 307, 121202. [Google Scholar] [CrossRef]
- Liu, W.; Li, L.; Lin, S.; Luo, Y.; Bao, Z.; Mao, Y.; Li, K.; Wu, D.; Peng, H. Confined Ni-In intermetallic alloy nanocatalyst with excellent coking resistance for methane dry reforming. J. Energy Chem. 2021, 65, 34–47. [Google Scholar] [CrossRef]
- Lu, Y.; Guo, D.; Zhao, Y.; Moyo, P.S.; Zhao, Y.; Wang, S.; Ma, X. Confined high dispersion of Ni nanoparticles derived from nickel phyllosilicate structure in silicalite-2 shell for dry reforming of methane with enhanced performance. Microporous Mesoporous Mater. 2020, 313, 110842. [Google Scholar] [CrossRef]
- Kong, W.; Fu, Y.; Shi, L.; Li, S.; Vovk, E.; Zhou, X.; Si, R.; Pan, B.; Yuan, C.; Li, S.; et al. Nickel nanoparticles with interfacial confinement mimic noble metal catalyst in methane dry reforming. Appl. Catal. B Environ. 2021, 285, 119837. [Google Scholar] [CrossRef]
- Xiao, M.; Wang, Z.; Lyu, M.; Luo, B.; Wang, S.; Liu, G.; Cheng, H.; Wang, L. Hollow Nanostructures for Photocatalysis: Advantages and Challenges. Adv. Mater. 2018, 31, e1801369. [Google Scholar] [CrossRef]
- Kosari, M.; Askari, S.; Seayad, A.M.; Xi, S.; Kawi, S.; Borgna, A.; Zeng, H.C. Strong coke-resistivity of spherical hollow Ni/SiO2 catalysts with shell-confined high-content Ni nanoparticles for methane dry reforming with CO2. Appl. Catal. B Environ. 2022, 310, 121360. [Google Scholar] [CrossRef]
- Wang, J.; Mao, Y.; Zhang, L.; Li, Y.; Liu, W.; Ma, Q.; Wu, D.; Peng, H. Remarkable basic-metal oxides promoted confinement catalysts for CO2 reforming. Fuel 2022, 315, 123167. [Google Scholar] [CrossRef]
- Park, K.S.; Goag, T.Y.; Kwon, J.H.; Park, Y.M.; Yu, J.S.; Jeong, H.E.; Choung, J.W.; Bae, J.W. Effects of spatially confined nickel nanoparticles in surface-pretreated hydrophobic SBA-15 for dry reforming of CH4 with CO2. J. CO2 Util. 2021, 51, 101629. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Fabrication Method | Reaction Conditions | SABET (m2 g−1) | Catalytic Performance | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Pressure (Bar) | Temperature (°C) | XCO2 (%) a | SCH4, (%) | STYCH4 Yields (μmolCH4⋅ gcat−1⋅s1) | ||||
| Ni7Fe@C | Pyrolysis Wet impregnation | 1 | 350 | 112.68 | 72.3 | 99.3 | - | [77] |
| Co/MnO@PGC | Pyrolysis | 1 | 240 | 163.50 | 32.1 | 99.6 | 13.34 | [78] |
| Ni/Al@Al2O3 | Hydrothermal surface oxidation (HTSO) | 1 | 500 | 172.00 | - | - | 7.21 | [82] |
| Co@mSiO2 Co@Silicalite-1 | Solvothermal | 1 | 400 | - | 56.3 43.4 | 80.4 48.2 | 48.5 20.3 | [60] |
| Ni@Silicalite-1 | Selective desilication via solvothermal | 1 | 450 | 367 | 38.3 | 60 | - | [83] |
| Ni-p@CeO2 (NPS-180-5C) | Hydrothermal | 1 | 450 | 18.2 | 75.0 | 92.0 | - | [81] |
| Ni/Al@MnAl2O4 Ni/Al@MgAl2O4 | Deposition–precipitation | 1 | 300 | 129 171 | 90 90 | 99 99 | - - | |
| Ni@MCM-41 Ni/0.05Mg@MCM-41 Ni/0.1Mg@MCM-41 | In situ hydrothermal | 10 | 350 | 622.5 606.3 498.5 | 89 84.3 80 | 90 95 97.8 | - | [87] |
| Ni@SiO2 | Stober | 15 | 450 | 263 | - | 89.8 | - | [88] |
| Ni@mpCeO2 | Solvothermal | - | 350 | 131 | 80 | 99 | - | |
| Catalysts | Fabrication Method | Reaction Conditions | SABET (m2⋅g−1) | Catalytic Performance | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Pressure (Bar) | Temp. (°C) | XCO2 (%) a | SMeOH, (%) | STYMeOH (gMeOH⋅gcat−1⋅h−1) | ||||
| Cu/ZnO@UiO-bpy | - | 40 | 250 | 117.8 | 3.3 | 100 | 2.59 | [96] |
| Cu@ZnOx Cu@ZnOx/ZnO | Coprecipitation | 30 | 250 | - - | 3 - | 100 100 | 4.6 146.0 | [99] |
| Cu@SiO2 In@SiO2 CuIn@SiO2 | Double-step solvothermal | 1 | 250 | 204.2 206.6 161.6 | 6.5 4.3 12.5 | 54.2 89.0 78.2 | 2.40 2.56 6.55 | [57] |
| Cu@mSiO2 CuZnO@mSiO2 | One pot Solvothermal | 50 | 250 | 618 589 | 10.2 9.8 | 26.5 66.6 | 56.6 136.6 | [100] |
| Cu-SiO2 phyllosilicate | Solvothermal | 1 | 225 | 159.7 | 3.5 | 77 | - | [94] |
| Hollow Cu@ZrO2 | Hydrothermal | 30 | 220 | 614.5 | 5 | 85 | 144 | [101] |
| Pd0.4@CuMgAlOX | Stober | 40 | 100 | 263 | - | 89.8 | - | [102] |
| Cu-ZnO@MVmSiO2 | Stober | 30 | 240 280 | - - | 23.0 34.0 | [103] | ||
| Cu-ZnO-Al2O3@MVmSiO2 | 30 | 240 280 | - - | 14.2 21.6 | ||||
| Cu-ZnO-ZrO2@MVmSiO2 | 30 | 240 280 | - - | 34.2 72.0 | ||||
| Catalysts | Fabrication Method | Reaction Conditions | Catalytic Performance | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|
| Pressure (Bar) | Temp. (°C) | XCO2 (%) | YCH4 (%) | Products H2/CO | Reaction Rates (molCH2/gcat/h) | Coke Form. (wt%) | |||
| 20% Ni@S-1 | Dissolution-recrystallization | 1 | 800 | 81.2 | <1.0 | 20.0 | 3.3 | [115] | |
| In0.5Ni@SiO2 | One pot microemulsion | 1 | 800 550 | 98 34 | 93 18 | 1.1 - | - - | - - | [116] |
| 5% Ni@S2-T | Hydrothermal | 1 | 700 800 | 75 - | 75 95 | 0.99 - | 40.2 - | 1.1 - | [117] |
| Ni@S-1 | Hydrothermal | 1 | 800 | - | 71 | - | - | 0 | [118] |
| Ni-HSs/SiO2 | Hydrothermal | 1 | 750 | 80 | 69 | 0.77 | - | 30 | [120] |
| 10Ni@CeO2/Al2O3 | EISA | 5 | 800 | 82 | 71 | 0.88 | - | 0.0 | [80] |
| Ni@Al2O3 | |||||||||
| Ni/MgO@DMS | Sol–gel | 1 | 800 550 | 96 38 | 88 35 | 0.99 0.69 | - - | 0 - | [121] |
| Ni/La2O3@DMS | So–gel | 1 | 550 | 40.5 | 35 | 0.71 | - | 1.13 | [122] |
| Ni(10)@SBA15 | Solvothermal | 1 | 800 | 83.1 | 73.5 | 0.87 | - | 0.9 | [122] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusdan, N.A.; Timmiati, S.N.; Isahak, W.N.R.W.; Yaakob, Z.; Lim, K.L.; Khaidar, D. Recent Application of Core-Shell Nanostructured Catalysts for CO2 Thermocatalytic Conversion Processes. Nanomaterials 2022, 12, 3877. https://doi.org/10.3390/nano12213877
Rusdan NA, Timmiati SN, Isahak WNRW, Yaakob Z, Lim KL, Khaidar D. Recent Application of Core-Shell Nanostructured Catalysts for CO2 Thermocatalytic Conversion Processes. Nanomaterials. 2022; 12(21):3877. https://doi.org/10.3390/nano12213877
Chicago/Turabian StyleRusdan, Nisa Afiqah, Sharifah Najiha Timmiati, Wan Nor Roslam Wan Isahak, Zahira Yaakob, Kean Long Lim, and Dalilah Khaidar. 2022. "Recent Application of Core-Shell Nanostructured Catalysts for CO2 Thermocatalytic Conversion Processes" Nanomaterials 12, no. 21: 3877. https://doi.org/10.3390/nano12213877