
Mesoscale Modeling of Phase Separation Controlled by Hydrosilylation in Polyhydromethylsiloxane (PHMS)-Containing Blends
 , ,
, , 
Abstract
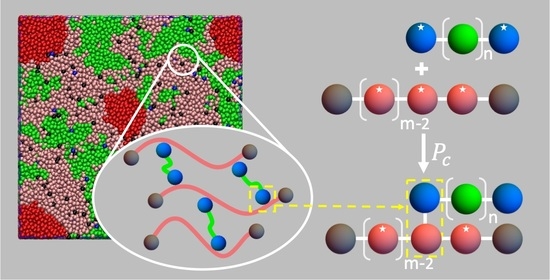
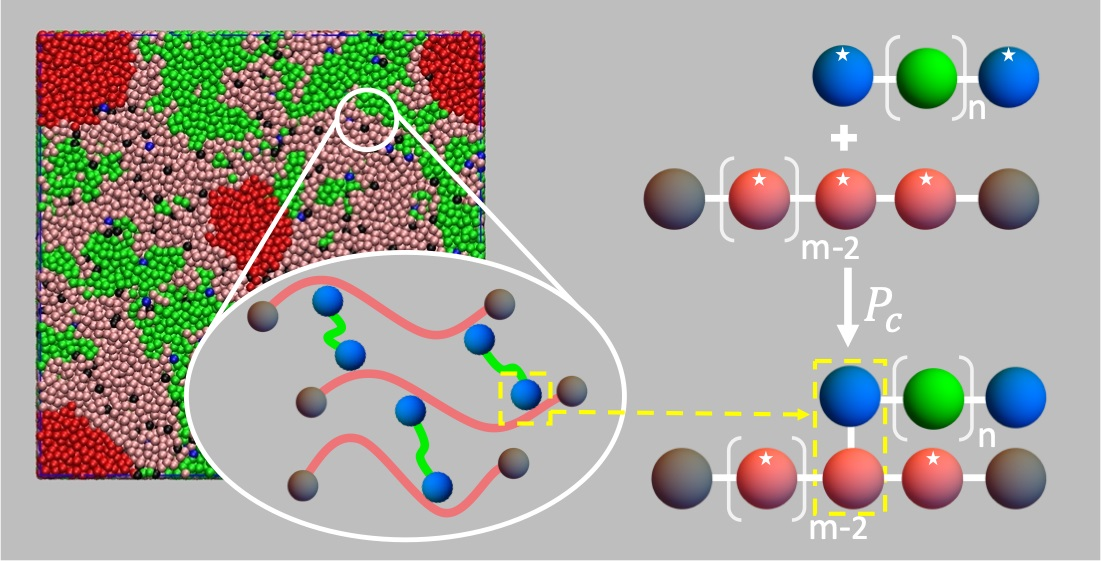
:
{kind=link}
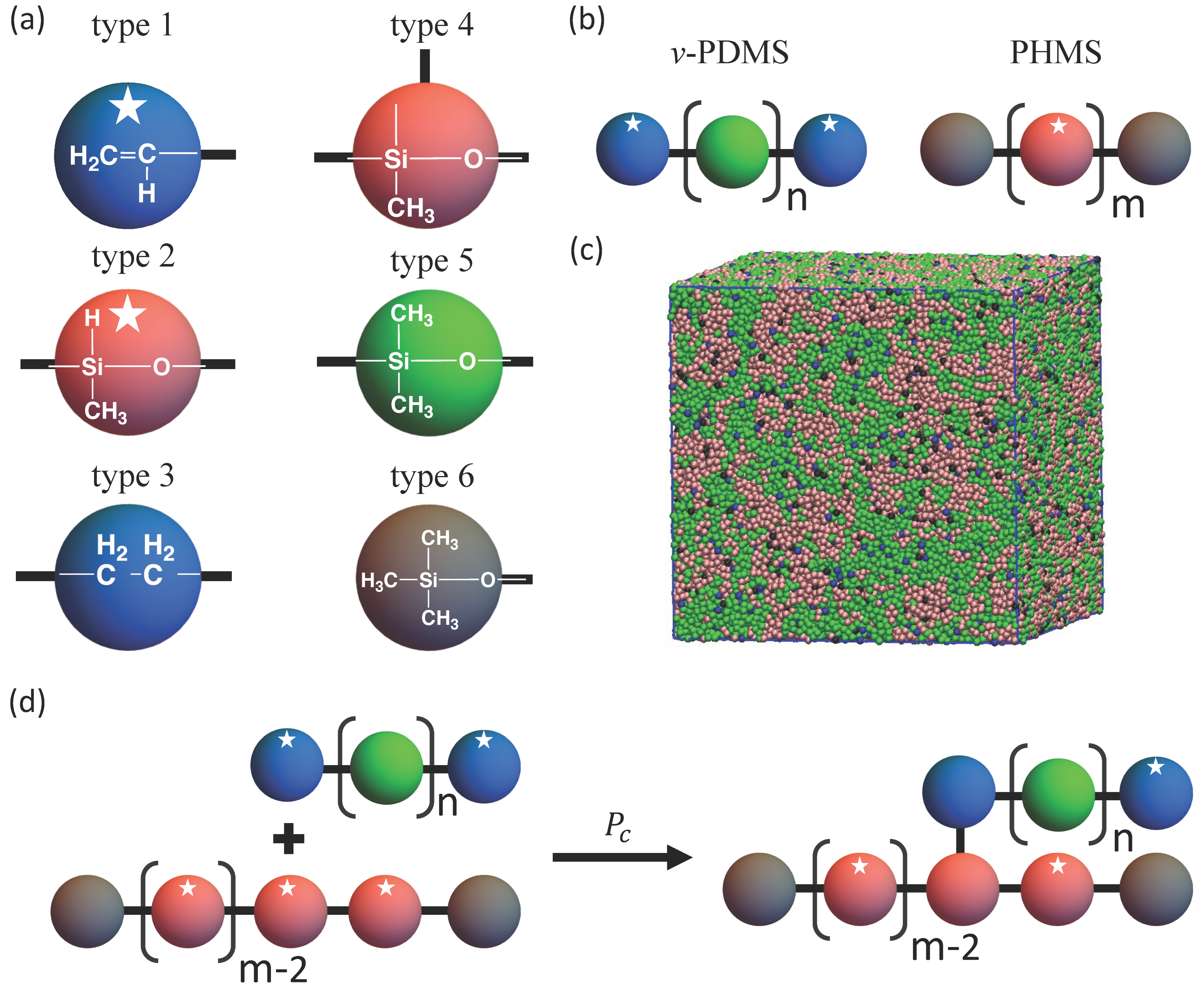{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Dissipative Particle Dynamics Approach
2.2. Defining Repulsion Parameters between Dissimilar Beads
2.3. Introducing Hydrosilylation Reaction within DPD Framework
3. Results
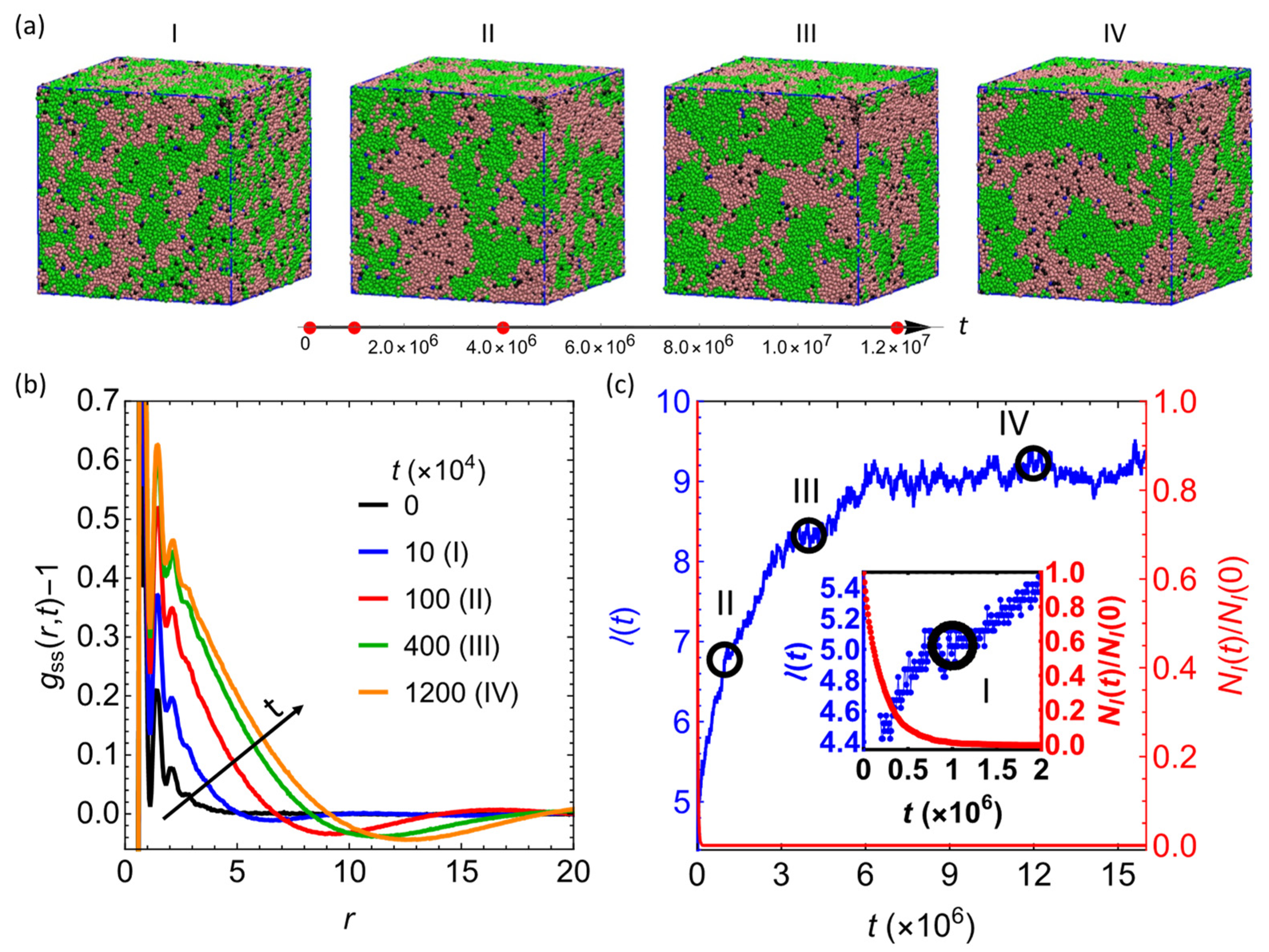
3.1. Hydrosilylation Reaction Arrests Domain Growth in Blends Containing PHMS
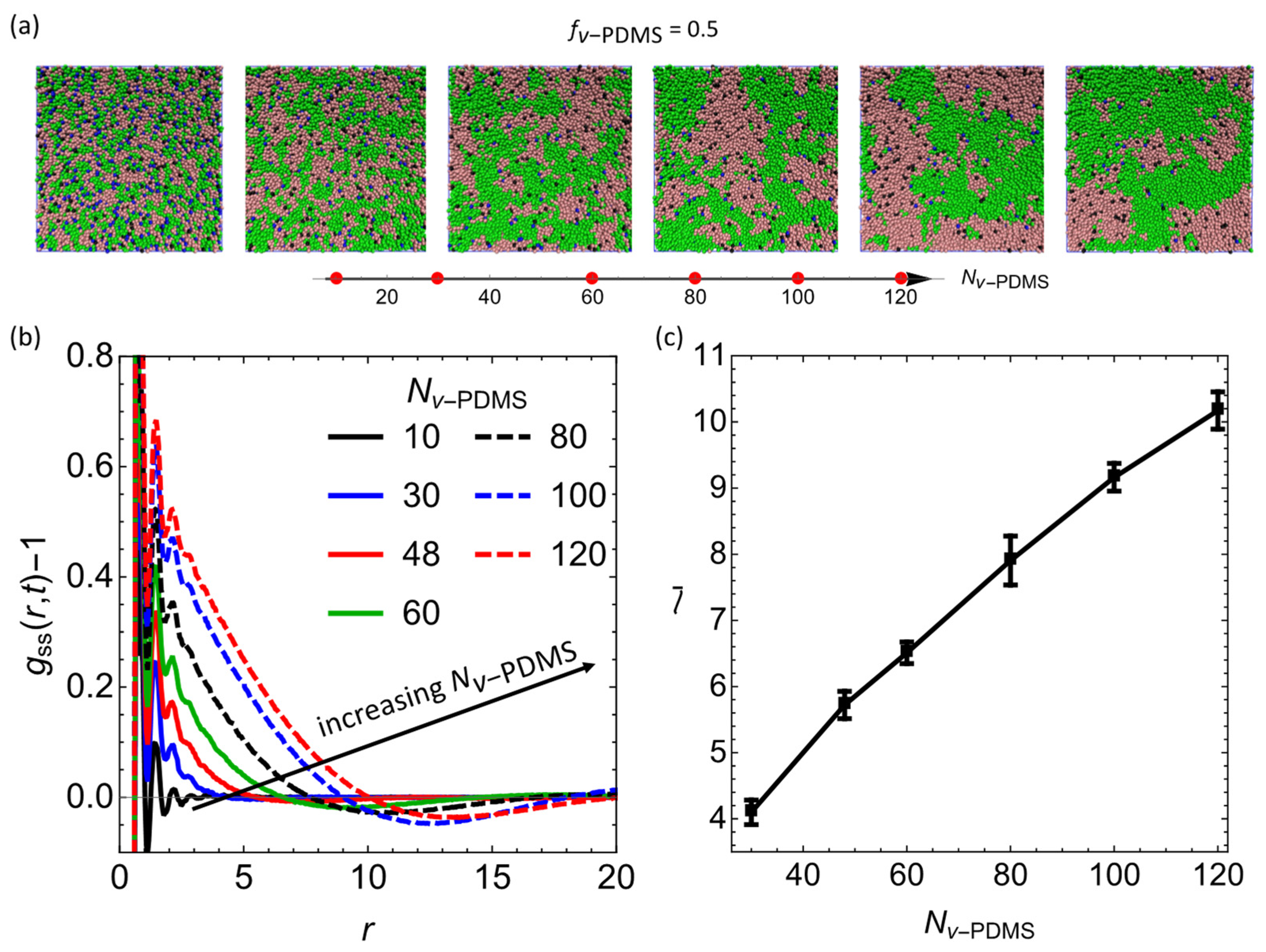
3.2. Effect of Degree of Polymerization of the Sacrificial Chains,
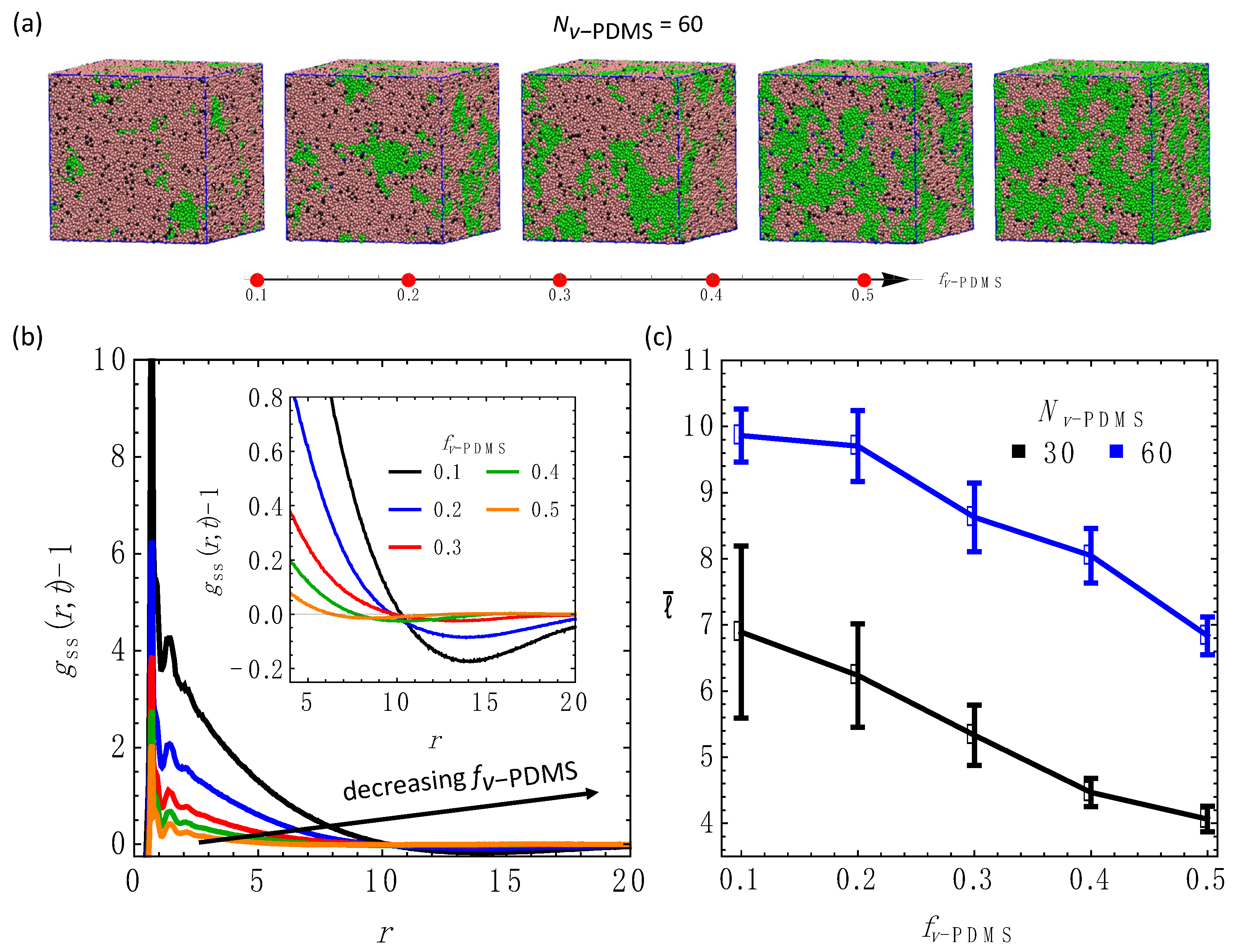
3.3. Effect of the Fraction of the Sacrificial Component,
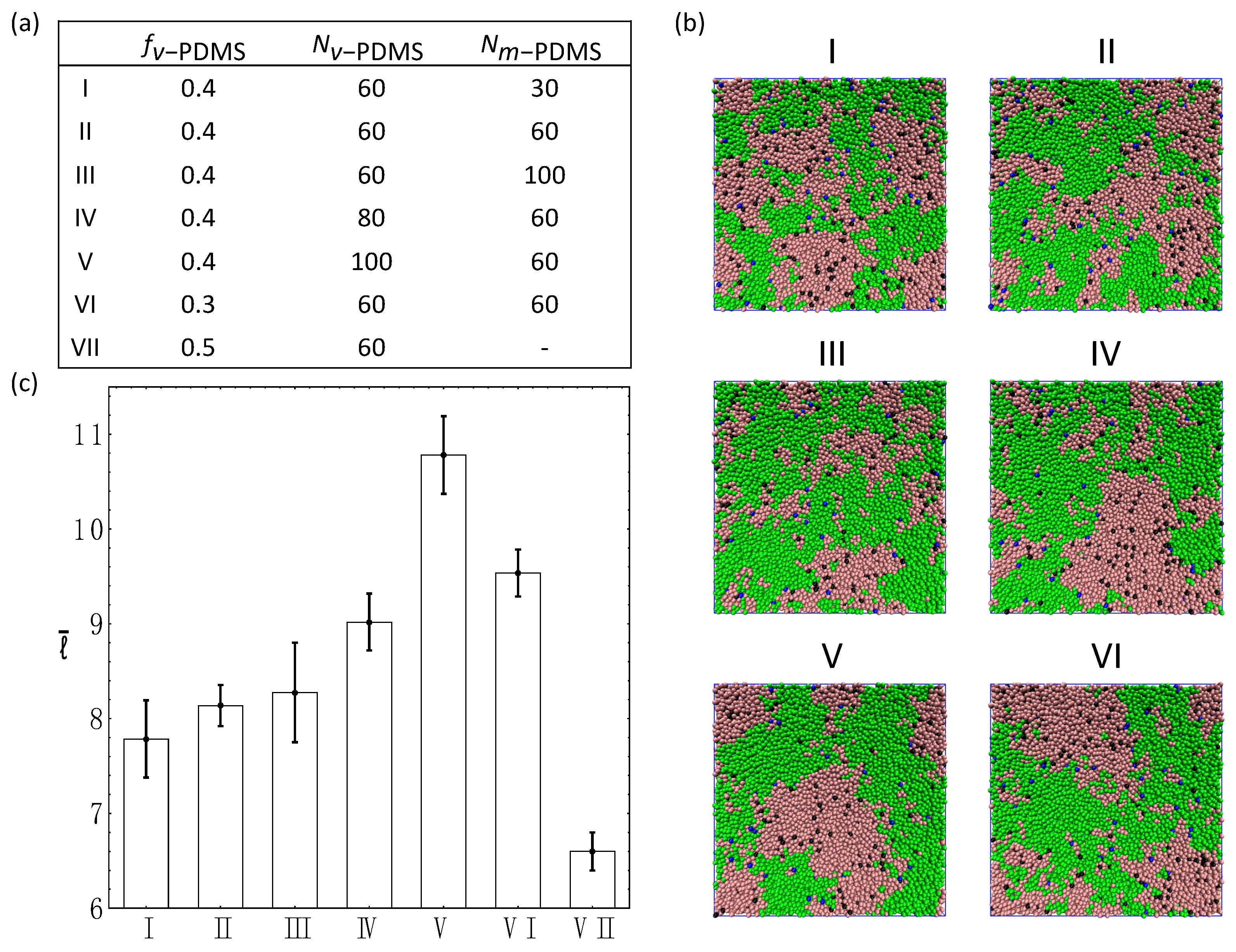
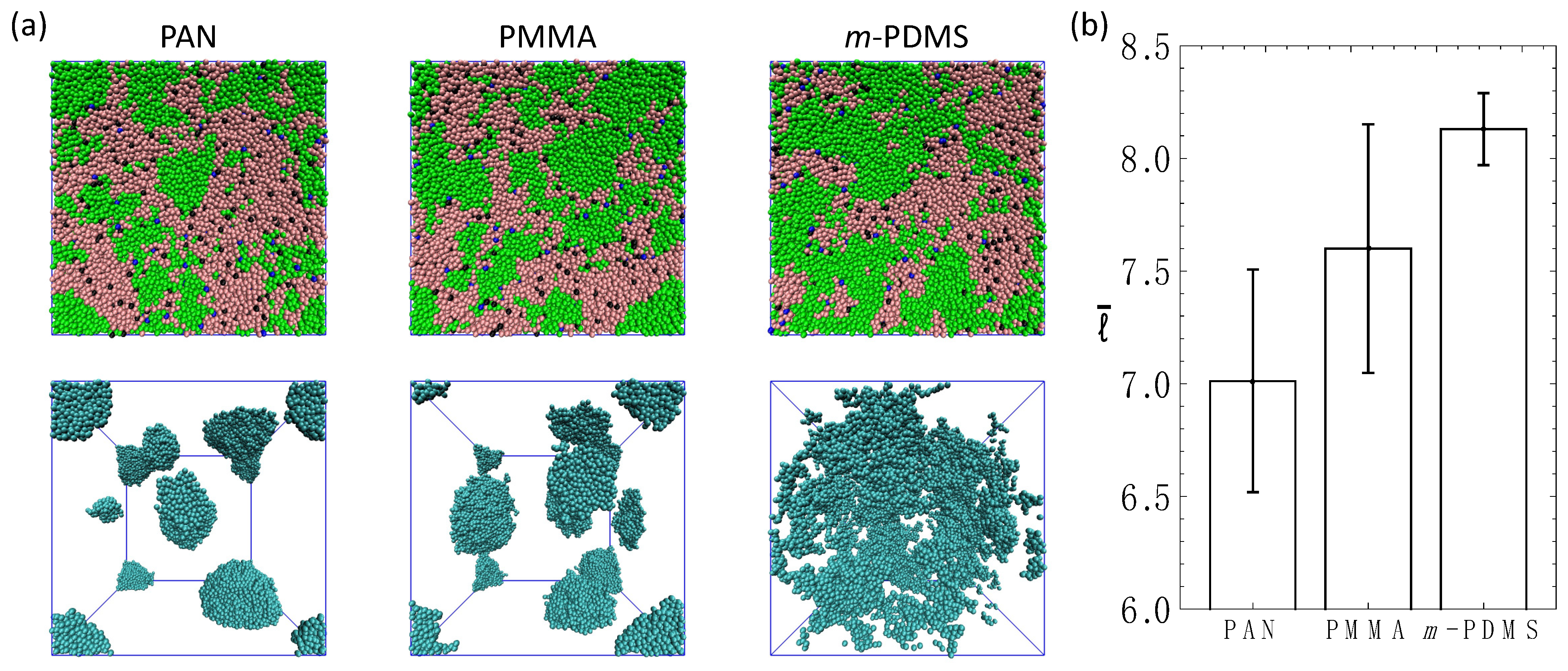
3.4. Ternary Systems Incorporating Reactive and Non-Reactive Sacrificial Components
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Z.W.; Bao, Y.P.; Sun, X.; Chen, K.; Zhou, M.J.; He, L.; Huang, Q.; Huang, Z.R.; Chai, Z.F.; Song, Y.J. Mesoporous Polymer-Derived Ceramic Membranes for Water Purification via a Self-Sacrificed Template. ACS Omega 2020, 5, 11100–11105. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.B.; Wang, F.H.; Yang, M.Y.; Yu, J.L.; Hao, L.Y.; Xu, X.; Wang, G.; Agathopoulos, S. Polymer-derived porous SiOC ceramic membranes for efficient oil-water separation and membrane distillation. J. Membr. Sci. 2019, 579, 111–119. [Google Scholar] [CrossRef]
- Chauhan, P.K.; Parameshwaran, R.; Kannan, P.; Madhavaram, R.; Sujith, R. Hydrogen storage in porous polymer derived SiliconOxycarbide ceramics: Outcomes and perspectives. Ceram. Int. 2021, 47, 2591–2599. [Google Scholar] [CrossRef]
- Li, J.K.; Lu, K. Highly Porous SiOC Bulk Ceramics with Water Vapor Assisted Pyrolysis. J. Am. Ceram. Soc. 2015, 98, 2357–2365. [Google Scholar] [CrossRef]
- Li, J.K.; Lu, K.; Lin, T.S.; Shen, F.Y. Preparation of Micro-/Mesoporous SiOC Bulk Ceramics. J. Am. Ceram. Soc. 2015, 98, 1753–1761. [Google Scholar] [CrossRef]
- Vakifahmetoglu, C.; Colombo, P. A direct method for the fabrication of macro-porous SiOC ceramics from preceramic polymers. Adv. Eng. Mater. 2008, 10, 256–259. [Google Scholar] [CrossRef]
- Colombo, P.; Mera, G.; Riedel, R.; Soraru, G.D. Polymer-Derived Ceramics: 40 Years of Research and Innovation in Advanced Ceramics. J. Am. Ceram. Soc. 2010, 93, 1805–1837. [Google Scholar] [CrossRef]
- Fu, S.Y.; Zhu, M.; Zhu, Y.F. Organosilicon polymer-derived ceramics: An overview. J. Adv. Ceram. 2019, 8, 457–478. [Google Scholar] [CrossRef]
- Vakifahmetoglu, C.; Zeydanli, D.; Colombo, P. Porous polymer derived ceramics. Mater. Sci. Eng. R 2016, 106, 1–30. [Google Scholar] [CrossRef]
- Abe, Y.; Gunji, T. Oligo- and polysiloxanes. Prog. Polym. Sci. 2004, 29, 149–182. [Google Scholar] [CrossRef]
- Kumar, B.V.M.; Kim, Y.W. Processing of polysiloxane-derived porous ceramics: A review. Sci. Technol. Adv. Mater. 2010, 11, 044303. [Google Scholar] [CrossRef]
- Dibandjo, P.; Dire, S.; Babonneau, F.; Soraru, G.D. Influence of the polymer architecture on the high temperature behavior of SiCO glasses: A comparison between linear- and cyclic-derived precursors. J. Non-Cryst. Solids 2010, 356, 132–140. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, C.; Cui, X.M.; Sun, J.H.; Ding, R.M.; Zhang, Q.H.; Xu, Y. Transparent and Dense Ladder-Like Alkylene-Bridged Polymethylsiloxane Coating with Enhanced Water Vapor Barrier Property. ACS Appl. Mater. Interfaces 2015, 7, 22157–22165. [Google Scholar] [CrossRef]
- Zalewski, K.; Chylek, Z.; Trzcinski, W.A. A Review of Polysiloxanes in Terms of Their Application in Explosives. Polymers 2021, 13, 1080. [Google Scholar] [CrossRef]
- Fortuniak, W.; Chojnowski, J.; Slomkowski, S.; Pospiech, P.; Kurjata, J. Route to hydrophilic, hydrophobic and functionalized cross-linked polysiloxane microspheres. Polymer 2013, 54, 3156–3165. [Google Scholar] [CrossRef]
- Greenough, M.; Zhao, Z.Y.; Jacobsohn, L.G.; Tong, J.H.; Bordia, R.K. Low/intermediate temperature pyrolyzed polysiloxane derived ceramics with increased carbon for electrical applications. J. Eur. Ceram. Soc. 2021, 41, 5882–5889. [Google Scholar] [CrossRef]
- Colombo, P. Engineering porosity in polymer-derived ceramics. J. Eur. Ceram. Soc. 2008, 28, 1389–1395. [Google Scholar] [CrossRef]
- Yu, Z.J.; Feng, Y.; Li, S.; Pei, Y.X. Influence of the polymer-polymer miscibility on the formation of mesoporous SiC(O) ceramics for highly efficient adsorption of organic dyes. J. Eur. Ceram. Soc. 2016, 36, 3627–3635. [Google Scholar] [CrossRef]
- Yan, X.; Su, D.; Han, S.S. Phase separation induced macroporous SiOC ceramics derived from polysiloxane. J. Eur. Ceram. Soc. 2015, 35, 443–450. [Google Scholar] [CrossRef]
- Wu, J.Q.; Li, Y.M.; Chen, L.M.; Zhang, Z.B.; Wang, D.; Xu, C.H. Simple fabrication of micro/nano-porous SiOC foam from polysiloxane. J. Mater. Chem. 2012, 22, 6542–6545. [Google Scholar] [CrossRef]
- Blum, Y.; Soraru, G.D.; Ramaswamy, A.P.; Hui, D.; Carturan, S.M. Controlled Mesoporosity in SiOC via Chemically Bonded Polymeric “Spacers”. J. Am. Ceram. Soc. 2013, 96, 2785–2792. [Google Scholar] [CrossRef]
- Erb, D.; Lu, K. Synthesis of SiOC using solvent-modified polymer precursors. Mater. Chem. Phys. 2019, 237, 121844. [Google Scholar] [CrossRef]
- Nakajima, Y.; Shimada, S. Hydrosilylation reaction of olefins: Recent advances and perspectives. RSC Adv. 2015, 5, 20603–20616. [Google Scholar] [CrossRef]
- Meister, T.K.; Riener, K.; Gigler, P.; Stohrer, J.; Herrmann, W.A.; Kuhn, F.E. Platinum Catalysis Revisited-Unraveling Principles of Catalytic Olefin Hydrosilylation. ACS Catal. 2016, 6, 1274–1284. [Google Scholar] [CrossRef]
- Hoogerbrugge, P.J.; Koelman, J. Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. Europhys. Lett. 1992, 19, 155–160. [Google Scholar] [CrossRef]
- Español, P.; Warren, P. Statistical Mechanics of Dissipative Particle Dynamics. Europhys. Lett. 1995, 30, 191–196. [Google Scholar] [CrossRef]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Espanol, P.; Warren, P.B. Perspective: Dissipative Particle Dynamics. J. Chem. Phys. 2017, 146, 150901. [Google Scholar] [CrossRef]
- Santo, K.P.; Neimark, A.V. Dissipative particle dynamics simulations in colloid and Interface science: A review. Adv. Colloid Interface Sci. 2021, 298, 102545. [Google Scholar] [CrossRef]
- Xu, Z.Y.; Yang, Y.; Zhu, G.L.; Chen, P.Y.; Huang, Z.H.; Dai, X.B.; Hou, C.L.; Yan, L.T. Simulating Transport of Soft Matter in Micro/Nano Channel Flows with Dissipative Particle Dynamics. Adv. Theory Simul. 2019, 2, 1800160. [Google Scholar] [CrossRef]
- Vishnyakov, A.; Lee, M.T.; Neimark, A.V. Prediction of the Critical Micelle Concentration of Nonionic Surfactants by Dissipative Particle Dynamics Simulations. J. Phys. Chem. Lett. 2013, 4, 797–802. [Google Scholar] [CrossRef]
- Mills, Z.G.; Mao, W.B.; Alexeev, A. Mesoscale modeling: Solving complex flows in biology and biotechnology. Trends Biotechnol. 2013, 31, 426–434. [Google Scholar] [CrossRef]
- Nikolov, S.V.; Fernandez-Nieves, A.; Alexeev, A. Behavior and mechanics of dense microgel suspensions. Proc. Natl. Acad. Sci. USA 2020, 117, 27096–27103. [Google Scholar] [CrossRef]
- Anakhov, M.V.; Gumerov, R.A.; Potemkin, I.I. Stimuli-responsive aqueous microgels: Properties and applications. Mendeleev Commun. 2020, 30, 555–562. [Google Scholar] [CrossRef]
- Chen, S.; Olson, E.; Jiang, S.; Yong, X. Nanoparticle assembly modulated by polymer chain conformation in composite materials. Nanoscale 2020, 12, 14560–14572. [Google Scholar] [CrossRef]
- Liu, H.; Li, M.; Lu, Z.Y.; Zhang, Z.G.; Sun, C.C. Influence of Surface-Initiated Polymerization Rate and Initiator Density on the Properties of Polymer Brushes. Macromolecules 2009, 42, 2863–2872. [Google Scholar] [CrossRef]
- Yong, X.; Kuksenok, O.; Matyjaszewski, K.; Balazs, A.C. Harnessing Interfacially-Active Nanorods to Regenerate Severed Polymer Gels. Nano Lett. 2013, 13, 6269–6274. [Google Scholar] [CrossRef]
- Yong, X.; Kuksenok, O.; Balazs, A.C. Modeling free radical polymerization using dissipative particle dynamics. Polymer 2015, 72, 217–225. [Google Scholar] [CrossRef]
- Singh, A.; Kuksenok, O.; Johnson, J.A.; Balazs, A.C. Tailoring the structure of polymer networks with iniferter-mediated photo-growth. Polym. Chem. 2016, 7, 2955–2964. [Google Scholar] [CrossRef]
- Singh, A.; Kuksenok, O.; Johnson, J.A.; Balazs, A.C. Photo-regeneration of severed gel with iniferter-mediated photo-growth. Soft Matter 2017, 13, 1978–1987. [Google Scholar] [CrossRef]
- Xu, D.; Ni, C.Y.; Zhu, Y.L.; Lu, Z.Y.; Xue, Y.H.; Liu, H. Kinetic step-growth polymerization: A dissipative particle dynamics simulation study. J. Chem. Phys. 2018, 148, 024901. [Google Scholar] [CrossRef]
- Berezkin, A.V.; Kudryavtsev, Y.V. Linear interfacial polymerization: Theory and simulations with dissipative particle dynamics. J. Chem. Phys. 2014, 141, 194906. [Google Scholar] [CrossRef]
- Liu, Y.; Aizenberg, J.; Balazs, A.C. Using Dissipative Particle Dynamics to Model Effects of Chemical Reactions Occurring within Hydrogels. Nanomaterials 2021, 11, 2764. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Chauhan, A.; Puri, S.; Singh, A. Photo-induced bond breaking during phase separation kinetics of block copolymer melts: A dissipative particle dynamics study. Soft Matter 2021, 17, 1802–1813. [Google Scholar] [CrossRef]
- Groot, R.D.; Rabone, K.L. Mesoscopic Simulation of Cell Membrane Damage, Morphology Change and Rupture by Nonionic Surfactants. Biophys. J. 2001, 81, 725–736. [Google Scholar] [CrossRef]
- Choudhury, C.K.; Palkar, V.; Kuksenok, O. Computational Design of Nanostructured Soft Interfaces: Focus on Shape Changes and Spreading of Cubic Nanogels. Langmuir 2020, 36, 7109–7123. [Google Scholar] [CrossRef]
- Sirk, T.W.; Slizoberg, Y.R.; Brennan, J.K.; Lisal, M.; Andzelm, J.W. An enhanced entangled polymer model for dissipative particle dynamics. J. Chem. Phys. 2012, 136, 134903. [Google Scholar] [CrossRef]
- LAMMPS Pair Style Srp/React. Available online: https://docs.lammps.org/pair_srp.html#pair-style-srp-react-command (accessed on 29 August 2022).
- Palkar, V.; Kuksenok, O. Controlling Degradation and Erosion of Polymer Networks: Insights from Mesoscale Modeling. J. Phys. Chem. B 2021, 126, 336–346. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- LAMMPS Molecular Dynamics Simulator. Available online: http://lammps.org (accessed on 15 July 2022).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graphics Modell. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wolfram Research, Inc. Mathematica; Version 12.0; Wolfram Research, Inc.: Champaign, IL, USA, 2019. [Google Scholar]
- Hiemenz, P.C.; Lodge, T.C. Polymer Chemistry; CRC Press: Boca Raton, FL, USA, 2007; p. 608. [Google Scholar]
- Barton, A.F.M. Handbook of Polymer-Liquid Interaction Parameters and Solubility Parameters; CRC Press: Boca Raton, FL, USA, 1990; p. 768. [Google Scholar]
- Utracki, L.A.; Wilkie, C.A. Polymer Blends Handbook; Springer: Dordrecht, The Netherlands, 2014. [Google Scholar]
- Sperling, L.H. Introduction to Physical Polymer Science; Wiley: Hoboken, NJ, USA, 2015; p. 880. [Google Scholar]
- Erb, D.; Lu, K. Influence of vinyl bonds from PDMS on the pore structure of polymer derived ceramics. Mater. Chem. Phys. 2018, 209, 217–226. [Google Scholar] [CrossRef]
- Simpson, T.R.E.; Parbhoo, B.; Keddie, J.L. The dependence of the rate of crosslinking in poly(dimethyl siloxane) on the thickness of coatings. Polymer 2003, 44, 4829–4838. [Google Scholar] [CrossRef]
- Simpson, T.R.E.; Tabatabaian, Z.; Jeynes, C.; Parbhoo, B.; Keddie, J.L. Influence of interfaces on the rates of crosslinking in poly(dimethyl siloxane) coatings. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 1421–1431. [Google Scholar] [CrossRef]
- Antic, V.V.; Antic, M.P.; Govedarica, M.N.; Dvornic, P.R. Kinetics and mechanism of the formation of poly[(1,1,3,3-tetramethyldisoxanyl)ethylene] and poly(methyidecylsiloxane) by hydrosilylation. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 2246–2258. [Google Scholar] [CrossRef]
- Hansen, J.P.; McDonald, I.R. Theory of Simple Liquids: With Applications to Soft Matter; Academic Press: San Diego, CA, USA, 2013. [Google Scholar]
- Bucior, K.; Yelash, L.; Binder, K. Phase separation of an asymmetric binary-fluid mixture confined in a nanoscopic slit pore: Molecular-dynamics simulations. Phys. Rev. E 2008, 77, 051602. [Google Scholar] [CrossRef]
- Yelash, L.; Virnau, P.; Paul, W.; Binder, K.; Muller, M. Spinodal decomposition of polymer solutions: A parallelized molecular dynamics simulation. Phys. Rev. E 2008, 78, 031801. [Google Scholar] [CrossRef]
- Liu, H.J.; Cavaliere, S.; Jones, D.J.; Roziere, J.; Paddison, S.J. Morphology of Hydrated Nafion through a Quantitative Cluster Analysis: A Case Study Based on Dissipative Particle Dynamics Simulations. J. Phys. Chem. C 2018, 122, 13130–13139. [Google Scholar] [CrossRef]
- Ma, W.J.; Maritan, A.; Banavar, J.R.; Koplik, J. Dynamics of Phase-Separation of Binary Fluids. Phys. Rev. A 1992, 45, R5347–R5350. [Google Scholar] [CrossRef]
- Sun, W.; Qian, C.X.; Mastronardi, M.L.; Wei, M.; Ozin, G.A. Hydrosilylation kinetics of silicon nanocrystals. Chem. Commun. 2013, 49, 11361–11363. [Google Scholar] [CrossRef]
- Lee, J.N.; Park, C.; Whitesides, G.M. Solvent compatibility of poly (dimethylsiloxane)-based microfluidic devices. Anal. Chem. 2003, 75, 6544–6554. [Google Scholar] [CrossRef]
- Mark, J.E. Physical Properties of Polymers Handbook; Springer: New York, NY, USA, 2007. [Google Scholar]
- Polymer Database. Available online: http://www.polymerdatabase.com (accessed on 15 July 2022).
- PDMS. Available online: https://polymerdatabase.com/polymers/Polydimethylsiloxane.html (accessed on 15 July 2022).
- PAN. Available online: https://polymerdatabase.com/polymers/polyacrylonitrile.html (accessed on 15 July 2022).
- PMMA. Available online: https://polymerdatabase.com/polymers/polymethylmethacrylate.html (accessed on 15 July 2022).
- Watanabe, H.; Miyauchi, T. Determination of solubility parameter for siloxane segment the solubility of iodine in liquid polydimethylsiloxane. J. Chem. Eng. Jpn. 1973, 6, 109–114. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, Y.; Choudhury, C.K.; Palkar, V.; Wunderlich, R.; Bordia, R.K.; Kuksenok, O. Mesoscale Modeling of Phase Separation Controlled by Hydrosilylation in Polyhydromethylsiloxane (PHMS)-Containing Blends. Nanomaterials 2022, 12, 3117. https://doi.org/10.3390/nano12183117
Xiong Y, Choudhury CK, Palkar V, Wunderlich R, Bordia RK, Kuksenok O. Mesoscale Modeling of Phase Separation Controlled by Hydrosilylation in Polyhydromethylsiloxane (PHMS)-Containing Blends. Nanomaterials. 2022; 12(18):3117. https://doi.org/10.3390/nano12183117
Chicago/Turabian StyleXiong, Yao, Chandan K. Choudhury, Vaibhav Palkar, Raleigh Wunderlich, Rajendra K. Bordia, and Olga Kuksenok. 2022. "Mesoscale Modeling of Phase Separation Controlled by Hydrosilylation in Polyhydromethylsiloxane (PHMS)-Containing Blends" Nanomaterials 12, no. 18: 3117. https://doi.org/10.3390/nano12183117