
Automated Quantum Dots Purification via Solid Phase Extraction

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Continuous Synthesis of CdSe/ZnS Core/Shell Nanoparticles
2.2. Purification of CdSe/ZnS Core/Shell Nanoparticles
2.2.1. Derivatization of SiO2 Beads
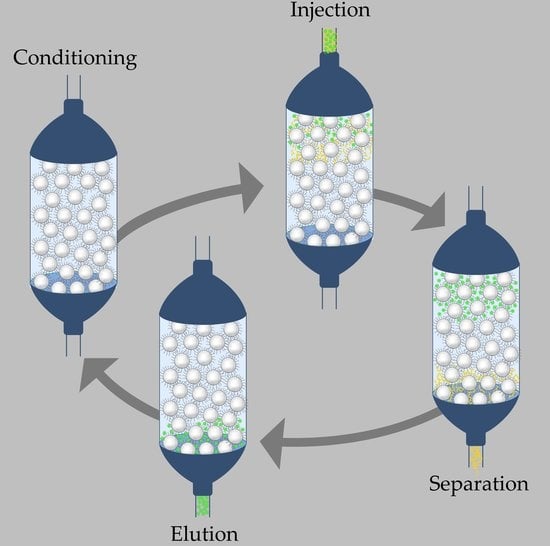
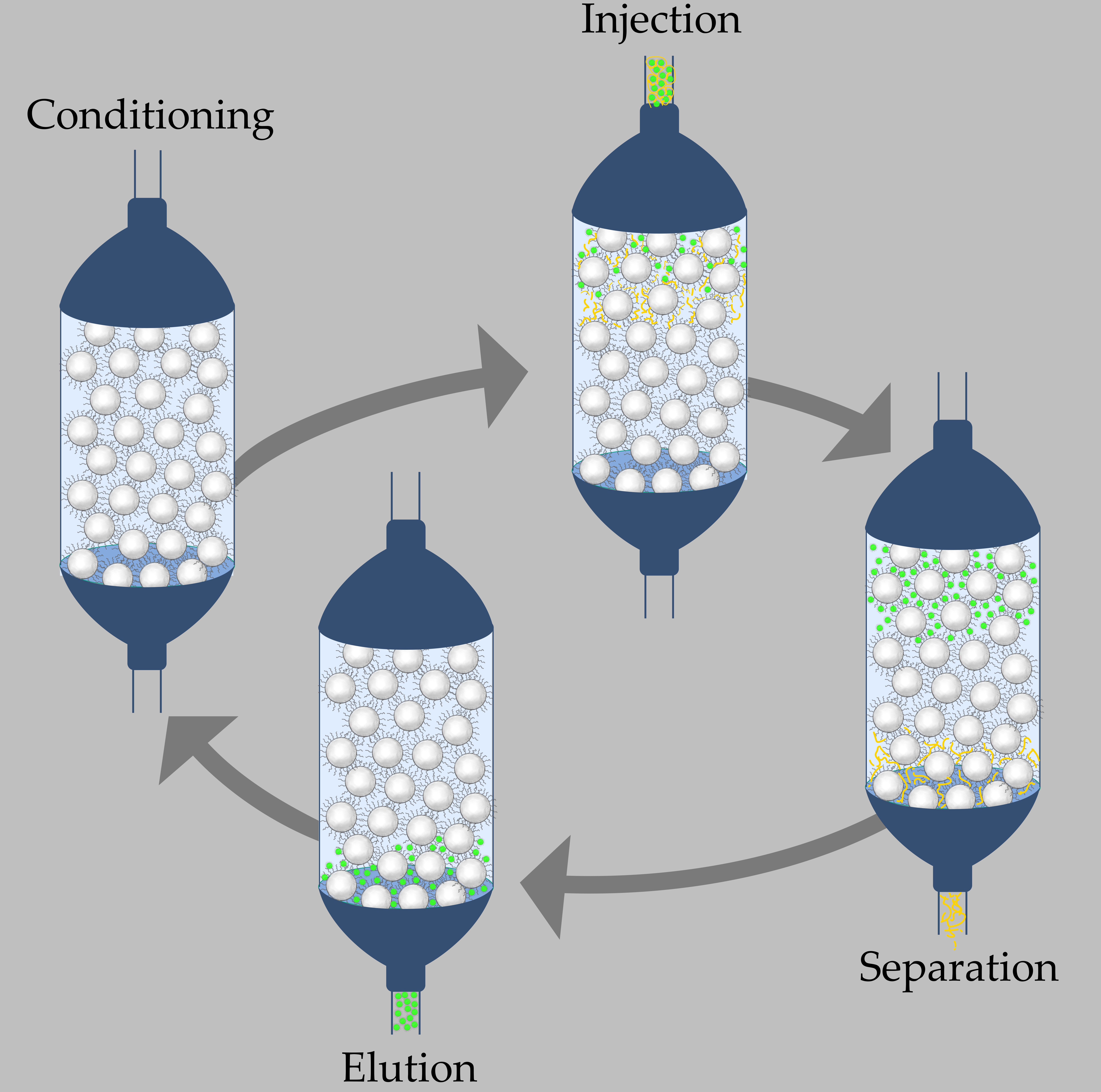
2.2.2. Purification Platform
2.2.3. Residence Time Distribution (VE)
2.3. Physicochemical Characterization
2.3.1. Photospectroscopical Determination of QDs Concentration and Fluorescence Intensity (QY)
2.3.2. Gas Chromatography and Mass Spectroscopy Analysis (GC/MS)
2.3.3. Thermogravimetric Measurements (TGA)
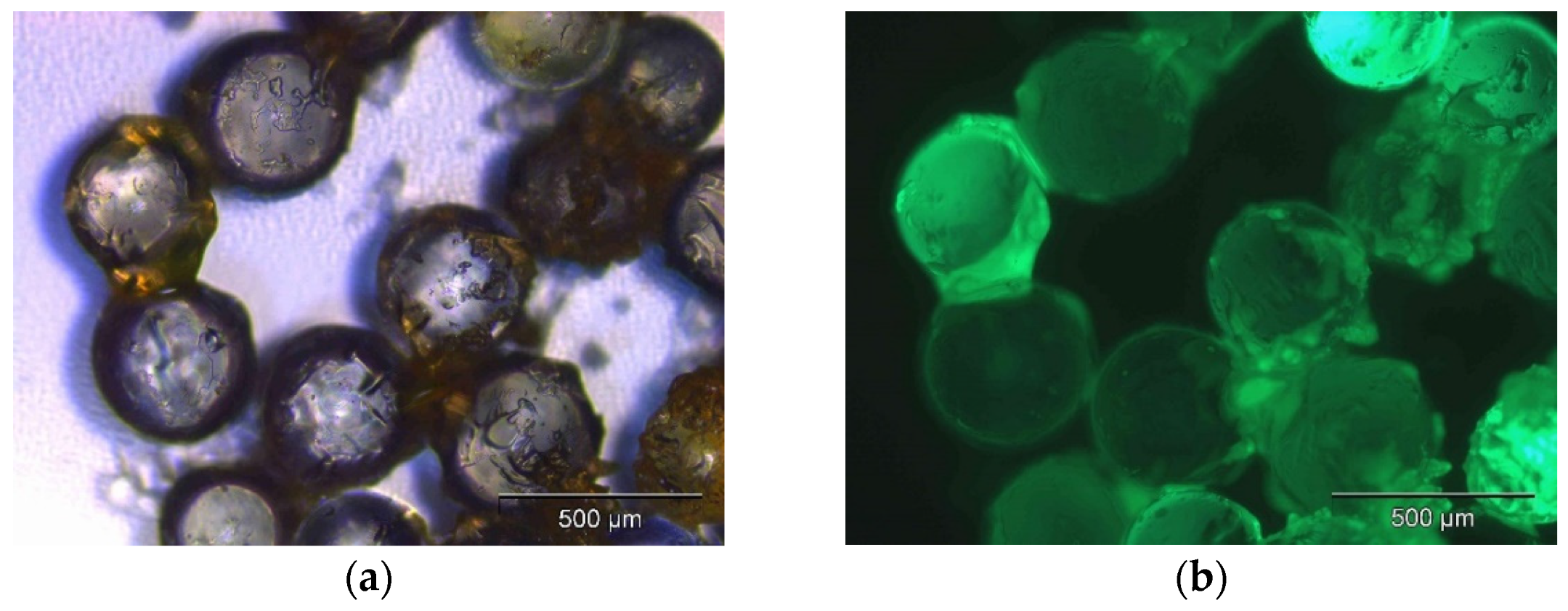
2.3.4. Scanning Electron Microscope (SEM)
2.3.5. Transmission Electron Microscopy (TEM)
2.4. Contact Angle
2.5. Hansen Solubility Parameter Analysis
Dynamic Light Scattering (DLS)
2.6. Analysis of Variance (ANOVA)
3. Results and Discussion
- Conditioning of the solid phase column with weak solvent
- Injection of crude product, i.e., QDs synthesis mixture and additional weak solvent, causing adhesion
- Injection of extraction solvent
- Elution of purified QDs via strong solvents.
3.1. Batch Assessment
3.2. Continuous Processing
3.2.1. Comparative Factor Analysis
3.2.2. Method Comparison
3.2.3. Hansen Solubility Parameter of QDs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Samsung. Business Report, 2020; Samsung Electronics: Suwon, Korea, 2021. [Google Scholar]
- Tomm, N.; Javadi, A.; Antoniadis, N.O.; Najer, D.; Löbl, M.C.; Korsch, A.R.; Schott, R.; Valentin, S.R.; Wieck, A.D.; Ludwig, A.; et al. A bright and fast source of coherent single photons. Nat. Nanotechnol. 2021, 16, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Resch-Genger, U.; Grabolle, M.; Cavaliere-Jaricot, S.; Nitschke, R.; Nann, T. Quantum dots versus organic dyes as fluorescent labels. Nat. Methods 2008, 5, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Jochum, T.; Ness, D.; Dieckmann, M.; Werner, K.; Niehaus, J.; Weller, H. Production and biofunctionalization of elongated semiconducting nanocrystals for ex-vivo applications. MRS Proc. 2014, 1635, 97–102. [Google Scholar] [CrossRef]
- Misuk, V.; Schmidt, M.; Braukmann, S.; Giannopoulos, K.; Karl, D.; Loewe, H. Segmented Flow-Based Multistep Synthesis of Cadmium Selenide Quantum Dots with Narrow Particle Size Distribution. Chem. Eng. Technol. 2015, 38, 1150–1153. [Google Scholar] [CrossRef]
- Kubendhiran, S.; Bao, Z.; Dave, K.; Liu, R.-S. Microfluidic Synthesis of Semiconducting Colloidal Quantum Dots and Their Applications. ACS Appl. Nano Mater. 2019, 2, 1773–1790. [Google Scholar] [CrossRef]
- Stavis, S.M.; Fagan, J.A.; Stopa, M.; Liddle, J.A. Nanoparticle Manufacturing—Heterogeneity through Processes to Products. ACS Appl. Nano Mater. 2018, 1, 4358–4385. [Google Scholar] [CrossRef]
- Shen, Y.; Gee, M.Y.; Greytak, A.B. Purification technologies for colloidal nanocrystals. Chem. Commun. 2017, 53, 827–841. [Google Scholar] [CrossRef]
- Menter, C.; Segets, D. Scalable classification of nanoparticles: A proof of principle for process design. Adv. Powder Technol. 2019, 30, 2801–2811. [Google Scholar] [CrossRef]
- Yang, Y.; Li, J.; Lin, L.; Peng, X. An efficient and surface-benign purification scheme for colloidal nanocrystals based on quantitative assessment. Nano Res. 2015, 8, 3353–3364. [Google Scholar] [CrossRef]
- Lim, H.; Woo, J.Y.; Lee, D.C.; Lee, J.; Jeong, S.; Kim, D. Continuous Purification of Colloidal Quantum Dots in Large-Scale Using Porous Electrodes in Flow Channel. Sci. Rep. 2017, 7, 43581. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.C.; Hendricks, M.P.; Choi, J.J.; Owen, J.S. Ligand exchange and the stoichiometry of metal chalcogenide nanocrystals: Spectroscopic observation of facile metal-carboxylate displacement and binding. J. Am. Chem. Soc. 2013, 135, 18536–18548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giansante, C.; Infante, I. Surface Traps in Colloidal Quantum Dots: A Combined Experimental and Theoretical Perspective. J. Phys. Chem. Lett. 2017, 8, 5209–5215. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Peng, X. Photogenerated excitons in plain core CdSe nanocrystals with unity radiative decay in single channel: The effects of surface and ligands. J. Am. Chem. Soc. 2015, 137, 4230–4235. [Google Scholar] [CrossRef]
- Grisorio, R.; Quarta, D.; Fiore, A.; Carbone, L.; Suranna, G.P.; Giansante, C. The dynamic surface chemistry of colloidal metal chalcogenide quantum dots. Nanoscale Adv. 2019, 1, 3639–3646. [Google Scholar] [CrossRef] [Green Version]
- Hassinen, A.; Moreels, I.; de Nolf, K.; Smet, P.F.; Martins, J.C.; Hens, Z. Short-chain alcohols strip X-type ligands and quench the luminescence of PbSe and CdSe quantum dots, acetonitrile does not. J. Am. Chem. Soc. 2012, 134, 20705–20712. [Google Scholar] [CrossRef] [PubMed]
- Giansante, C. Library Design of Ligands at the Surface of Colloidal Nanocrystals. Acc. Chem. Res. 2020, 53, 1458–1467. [Google Scholar] [CrossRef]
- Shen, Y.; Roberge, A.; Tan, R.; Gee, M.Y.; Gary, D.C.; Huang, Y.; Blom, D.A.; Benicewicz, B.C.; Cossairt, B.M.; Greytak, A.B. Gel permeation chromatography as a multifunctional processor for nanocrystal purification and on-column ligand exchange chemistry. Chem. Sci. 2016, 7, 5671–5679. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Weeranoppanant, N.; Xie, L.; Chen, Y.; Lusardi, M.R.; Imbrogno, J.; Bawendi, M.G.; Jensen, K.F. Multistage extraction platform for highly efficient and fully continuous purification of nanoparticles. Nanoscale 2017, 9, 7703–7707. [Google Scholar] [CrossRef]
- Hansen, C.M. Hansen Solubility Parameters: A User’s Handbook, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007; ISBN 9780849372483. [Google Scholar]
- Hansen, C.M. The Three Dimensional Solubility Parameter and Solvent Diffusion Coefficient, Their Importance in Surface Coating Formulation; Danish Technical Press: Copenhagen, Denmark, 1967. [Google Scholar]
- Süß, S.; Sobisch, T.; Peukert, W.; Lerche, D.; Segets, D. Determination of Hansen parameters for particles: A standardized routine based on analytical centrifugation. Adv. Powder Technol. 2018, 29, 1550–1561. [Google Scholar] [CrossRef]
- Abbott, S.; Holmes, N. Nanocoatings: Principles and Practice: From Research to Production; DEStech Publications Inc.: Lancaster, PA, USA, 2013; ISBN 1605950904. [Google Scholar]
- Bapat, S.; Kilian, S.O.; Wiggers, H.; Segets, D. Towards a framework for evaluating and reporting Hansen solubility parameters: Applications to particle dispersions. Nanoscale Adv. 2021, 3, 4400–4410. [Google Scholar] [CrossRef]
- Schindler, C. Einfluss der Kontinuierlichen Prozessführung auf die Bildung und die Eigenschaften von Nanopartikeln; Johannes Gutenberg-Universität: Mainz, Germany, 2022. [Google Scholar]
- Schindler, C.; Sperling, R.A. Process Control for the Continuous Synthesis of Quantum Dots via Inline Process Analytics. Chem. Ing. Tech. 2016, 88, 1224–1225. [Google Scholar] [CrossRef]
- Löb, P.; Drese, K.S.; Hessel, V.; Hardt, S.; Hofmann, C.; Löwe, H.; Schenk, R.; Schönfeld, F.; Werner, B. Steering of Liquid Mixing Speed in Interdigital Micro Mixers—From Very Fast to Deliberately Slow Mixing. Chem. Eng. Technol. 2004, 27, 340–345. [Google Scholar] [CrossRef]
- Hessel, V.; Hardt, S.; Löwe, H.; Schönfeld, F. Laminar mixing in different interdigital micromixers: I. Experimental characterization. AIChE J. 2003, 49, 566–577. [Google Scholar] [CrossRef]
- Löb, P. New Microreactor Designs for Practical Applications Realized by Additive Manufacturing. In Flow Chemistry: Integrated Approaches for Practical Applications; Luis, S.V., García-Verdugo, E., Eds.; Royal Society of Chemistry: Cambridge, UK, 2020; Chapter 12; pp. 388–415. ISBN 978-1-78801-498-4. [Google Scholar]
- Arkles, B. Hydrophobicity, Hydrophilicity and Silane Surface Modification; Gelest Inc.: Morrisville, NC, USA, 2011. [Google Scholar]
- Lecot, S.; Lavigne, A.; Yang, Z.; Géhin, T.; Botella, C.; Jousseaume, V.; Chevolot, Y.; Phaner-Goutorbe, M.; Yeromonahos, C. Arrangement of Monofunctional Silane Molecules on Silica Surfaces: Influence of Alkyl Chain Length, Head-Group Charge, and Surface Coverage, from Molecular Dynamics Simulations, X-ray Photoelectron Spectroscopy, and Fourier Transform Infrared Spectroscopy. J. Phys. Chem. C 2020, 124, 20125–20134. [Google Scholar] [CrossRef]
- Würth, C.; Grabolle, M.; Pauli, J.; Spieles, M.; Resch-Genger, U. Relative and absolute determination of fluorescence quantum yields of transparent samples. Nat. Protoc. 2013, 8, 1535–1550. [Google Scholar] [CrossRef] [PubMed]
- Sachs, L. Applied Statistics: A Handbook of Techniques, 2nd ed.; Springer: New York, NY, USA, 1984; ISBN 9781461252467. [Google Scholar]
- Jasieniak, J.; Smith, L.; van Embden, J.; Mulvaney, P.; Califano, M. Re-examination of the Size-Dependent Absorption Properties of CdSe Quantum Dots. J. Phys. Chem. C Nanomater. Interfaces 2009, 113, 19468–19474. [Google Scholar] [CrossRef]
- Dover, M.V.; Hensley, W.A. Properties of 1-Octadecene, n-Octadecane, and Di-m-tolylethane. Ind. Eng. Chem. 1935, 27, 337–339. [Google Scholar] [CrossRef]
- Moumouzias, G.; Ritzoulis, G. Viscosities and densities for propylene carbonate + toluene at 15, 20, 25, 30, and 35 °C. J. Chem. Eng. Data 1992, 37, 482–483. [Google Scholar] [CrossRef]
- Design-Expert Software, version #12; Stat-Ease Incorporation: Minneapolis, MN, USA, 2019.
- Abbott, S. Sticking Together: The Science of Adhesion; Royal Society of Chemistry: London, UK, 2020; ISBN 9781788018043. [Google Scholar]
- Díaz de los Ríos, M.; Hernández Ramos, E. Determination of the Hansen solubility parameters and the Hansen sphere radius with the aid of the solver add-in of Microsoft Excel. SN Appl. Sci. 2020, 2, 676. [Google Scholar] [CrossRef] [Green Version]
- Howell, J.; Roesing, M.; Boucher, D. A Functional Approach to Solubility Parameter Computations. J. Phys. Chem. B 2017, 121, 4191–4201. [Google Scholar] [CrossRef]
- Schreuder, M.A.; Gosnell, J.D.; Smith, N.J.; Warnement, M.R.; Weiss, S.M.; Rosenthal, S.J. Encapsulated white-light CdSe nanocrystals as nanophosphors for solid-state lighting. J. Mater. Chem. 2008, 18, 970. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sorbent | Hydrophilic | Hydrophobic | Perfluorinated | |||
|---|---|---|---|---|---|---|
| Solvent | ODE | ODE + OLA | ODE | ODE + OLA | ODE | ODE + OLA |
| ACN | 71° | 21° | 45° | 22° | 91° | 58° |
| MeOH | No contact | 73° | 61° | 47° | 90° | 109° |
| EtOH | No contact | 70° | 65° | 58° | No contact | No contact |
| Solvent | Amount | Sorbent | Ø/μm |
|---|---|---|---|
| MeOH | 1 × VE | Hydrocarbon | 70–110 |
| ACN | 2 × VE | Perfluorocarbon | 250–500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lüdicke, M.G.; Hildebrandt, J.; Schindler, C.; Sperling, R.A.; Maskos, M. Automated Quantum Dots Purification via Solid Phase Extraction. Nanomaterials 2022, 12, 1983. https://doi.org/10.3390/nano12121983
Lüdicke MG, Hildebrandt J, Schindler C, Sperling RA, Maskos M. Automated Quantum Dots Purification via Solid Phase Extraction. Nanomaterials. 2022; 12(12):1983. https://doi.org/10.3390/nano12121983
Chicago/Turabian StyleLüdicke, Malín G., Jana Hildebrandt, Christoph Schindler, Ralph A. Sperling, and Michael Maskos. 2022. "Automated Quantum Dots Purification via Solid Phase Extraction" Nanomaterials 12, no. 12: 1983. https://doi.org/10.3390/nano12121983