
Free Energy Surfaces and Barriers for Vacancy Diffusion on Al(100), Al(110), Al(111) Reconstructed Surfaces

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
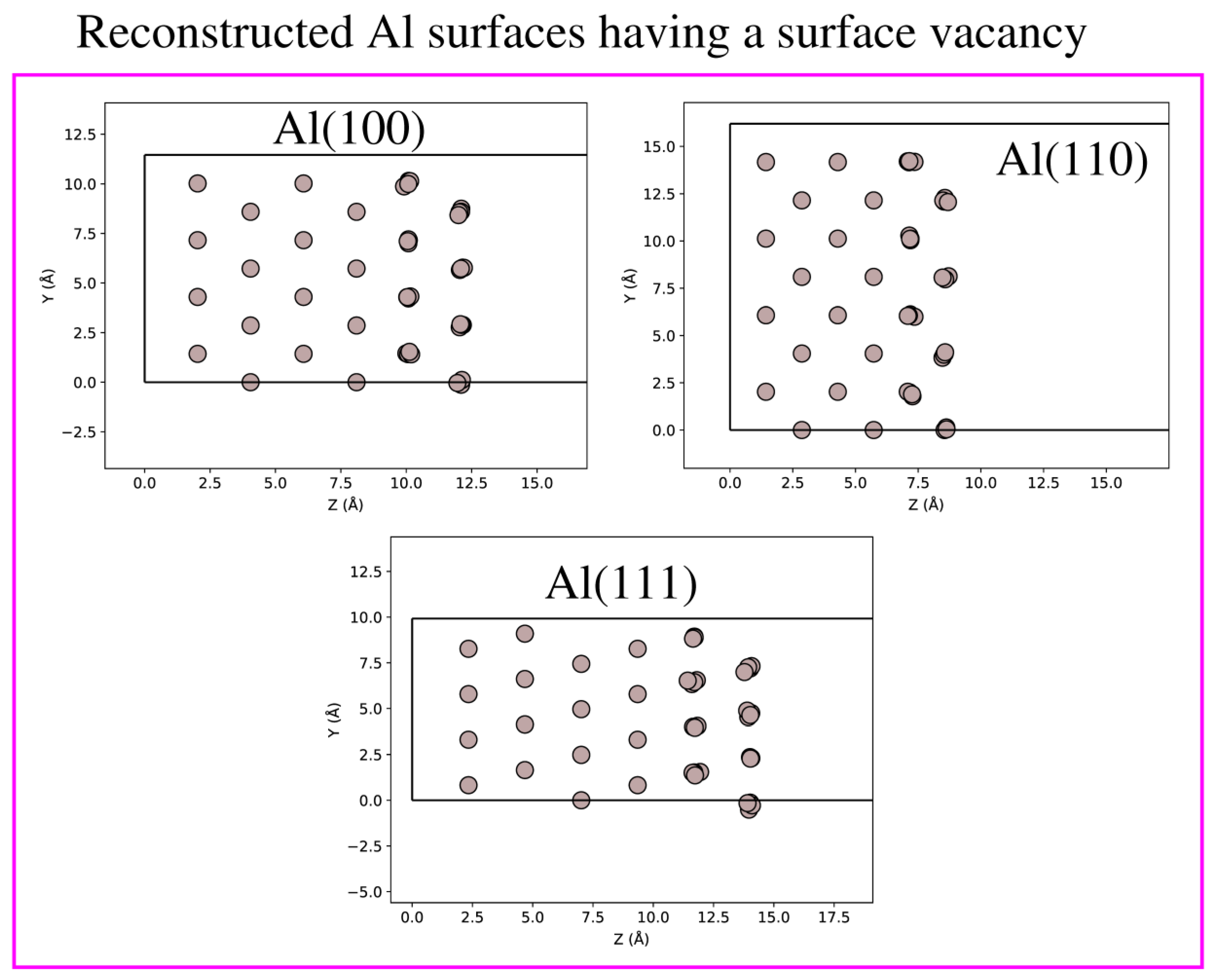
2. System and Computational Aspects
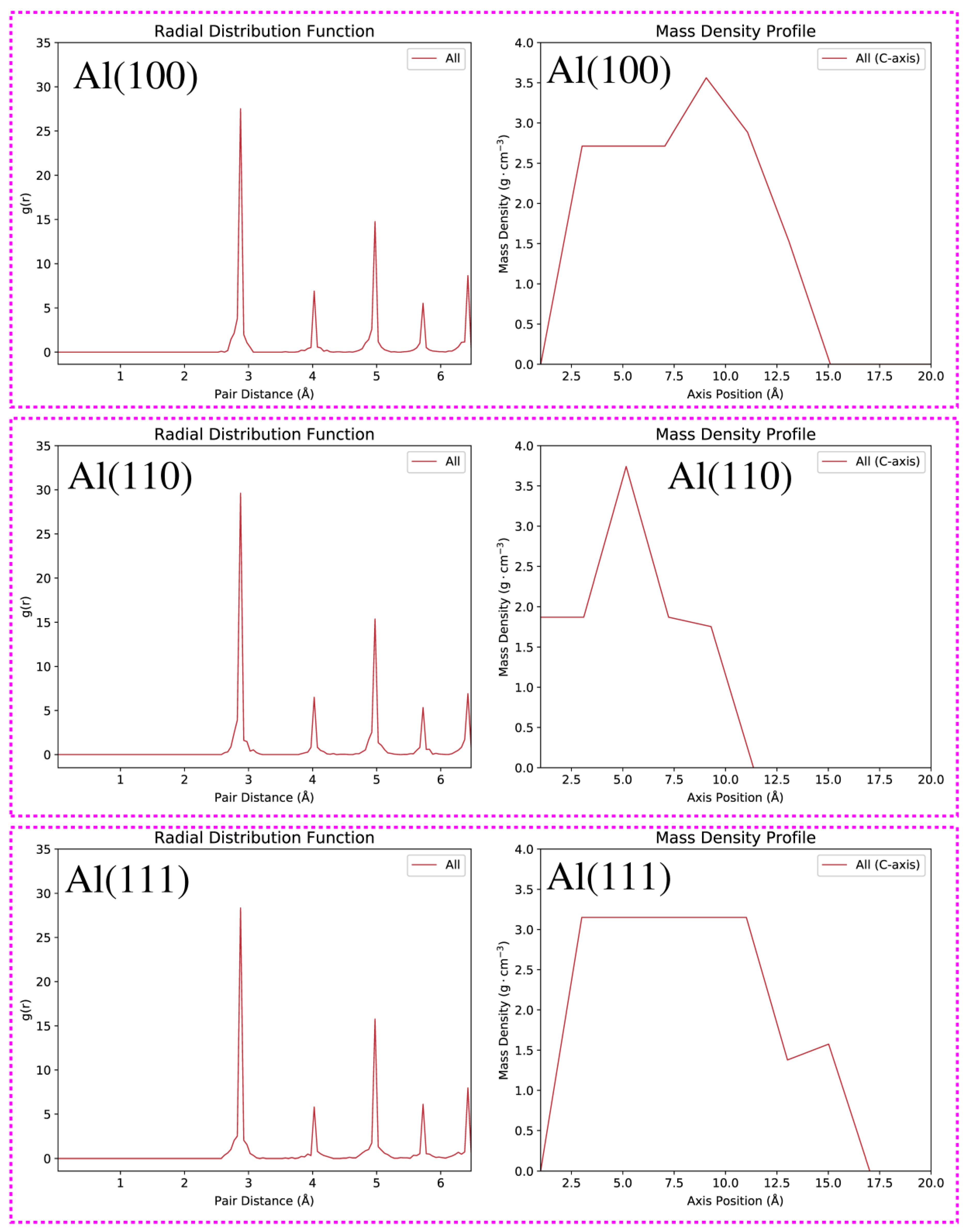
3. Results and Discussion
4. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Nomenclature
| Å | Angstrom |
| CV | Collective variable |
| EAM | Embedded atom method |
| F(s) | Free energy surface |
| MetaD | Metadynamic |
| ns | Nanosecond |
| RDF | Radial distribution function |
References
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How Fast-Folding Proteins Fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Tiwary, P.; Limongelli, V.; Salvalaglio, M.; Parrinello, M. Kinetics of protein–ligand unbinding: Predicting pathways, rates, and rate-limiting steps. Proc. Natl. Acad. Sci. USA 2015, 112, E386–E391. [Google Scholar] [CrossRef] [Green Version]
- Torrie, G.; Valleau, J. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Grubmüller, H. Predicting slow structural transitions in macromolecular systems: Conformational flooding. Phys. Rev. E 1995, 52, 2893–2906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprik, M.; Ciccotti, G. Free energy from constrained molecular dynamics. J. Chem. Phys. 1998, 109, 7737–7744. [Google Scholar] [CrossRef]
- Voter, A.F. A method for accelerating the molecular dynamics simulation of infrequent events. J. Chem. Phys. 1997, 106, 4665–4677. [Google Scholar] [CrossRef]
- Steiner, M.M.; Genilloud, P.A.; Wilkins, J.W. Simple bias potential for boosting molecular dynamics with the hyperdynamics scheme. Phys. Rev. B 1998, 57, 10236–10239. [Google Scholar] [CrossRef] [Green Version]
- VandeVondele, J.; Rothlisberger, U. Efficient multidimensional free energy calculations for ab initio molecular dynamics using classical bias potentials. J. Chem. Phys. 2000, 113, 4863–4868. [Google Scholar] [CrossRef]
- Rahman, J.A.; Tully, J.C. Puddle-skimming: An efficient sampling of multidimensional configuration space. J. Chem. Phys. 2002, 116, 8750–8760. [Google Scholar] [CrossRef]
- Mezei, M. Adaptive umbrella sampling: Self-consistent determination of the non-Boltzmann bias. J. Comput. Phys. 1987, 68, 237–248. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- El-Mellouhi, F.; Mousseau, N.; Lewis, L.J. Kinetic activation-relaxation technique: An off-lattice self-learning kinetic Monte Carlo algorithm. Phys. Rev. B 2008, 78, 153202. [Google Scholar] [CrossRef] [Green Version]
- Mees, M.J.; Pourtois, G.; Neyts, E.C.; Thijsse, B.J.; Stesmans, A. Uniform-acceptance force-bias Monte Carlo method with time scale to study solid-state diffusion. Phys. Rev. B 2012, 85, 134301. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Valsson, O.; Tiwary, P.; Parrinello, M.; Lindorff-Larsen, K. Frequency adaptive metadynamics for the calculation of rare-event kinetics. J. Chem. Phys. 2018, 149, 072309. [Google Scholar] [CrossRef]
- Debnath, J.; Parrinello, M. Gaussian Mixture-Based Enhanced Sampling for Statics and Dynamics. J. Phys. Chem. Lett. 2020, 11, 5076–5080. [Google Scholar] [CrossRef]
- Bartels, C.; Karplus, M. Probability Distributions for Complex Systems: Adaptive Umbrella Sampling of the Potential Energy. J. Phys. Chem. B 1998, 102, 865–880. [Google Scholar] [CrossRef]
- Maragliano, L.; Vanden-Eijnden, E. A temperature accelerated method for sampling free energy and determining reaction pathways in rare events simulations. Chem. Phys. Lett. 2006, 426, 168–175. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. WIREs Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Sutto, L.; Marsili, S.; Gervasio, F.L. New advances in metadynamics. WIREs Comput. Mol. Sci. 2012, 2, 771–779. [Google Scholar] [CrossRef]
- Valsson, O.; Tiwary, P.; Parrinello, M. Enhancing Important Fluctuations: Rare Events and Metadynamics from a Conceptual Viewpoint. Annu. Rev. Phys. Chem. 2016, 67, 159–184. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Laio, A.; Parrinello, M. Equilibrium Free Energies from Nonequilibrium Metadynamics. Phys. Rev. Lett. 2006, 96, 090601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaendtner, J.; Bonomi, M. Efficient Sampling of High-Dimensional Free-Energy Landscapes with Parallel Bias Metadynamics. J. Chem. Theory Comput. 2015, 11, 5062–5067. [Google Scholar] [CrossRef] [PubMed]
- Branduardi, D.; Bussi, G.; Parrinello, M. Metadynamics with Adaptive Gaussians. J. Chem. Theory Comput. 2012, 8, 2247–2254. [Google Scholar] [CrossRef]
- Casasnovas, R.; Limongelli, V.; Tiwary, P.; Carloni, P.; Parrinello, M. Unbinding Kinetics of a p38 MAP Kinase Type II Inhibitor from Metadynamics Simulations. J. Am. Chem. Soc. 2017, 139, 4780–4788. [Google Scholar] [CrossRef]
- Dama, J.F.; Rotskoff, G.; Parrinello, M.; Voth, G.A. Transition-Tempered Metadynamics: Robust, Convergent Metadynamics via On-the-Fly Transition Barrier Estimation. J. Chem. Theory Comput. 2014, 10, 3626–3633. [Google Scholar] [CrossRef]
- Stumpf, R.; Scheffler, M. Ab initio calculations of energies and self-diffusion on flat and stepped surfaces of Al and their implications on crystal growth. Phys. Rev. B 1996, 53, 4958–4973. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Ferguson, A.L. Molecular enhanced sampling with autoencoders: On-the-fly collective variable discovery and accelerated free energy landscape exploration. J. Comput. Chem. 2018, 39, 2079–2102. [Google Scholar] [CrossRef] [Green Version]
- Fiorin, G.; Klein, M.L.; Hénin, J. Using collective variables to drive molecular dynamics simulations. Mol. Phys. 2013, 111, 3345–3362. [Google Scholar] [CrossRef]
- Bonati, L.; Rizzi, V.; Parrinello, M. Data-Driven Collective Variables for Enhanced Sampling. J. Phys. Chem. Lett. 2020, 11, 2998–3004. [Google Scholar] [CrossRef] [PubMed]
- Mendels, D.; Piccini, G.; Parrinello, M. Collective Variables from Local Fluctuations. J. Phys. Chem. Lett. 2018, 9, 2776–2781. [Google Scholar] [CrossRef] [PubMed]
- Brotzakis, Z.F.; Limongelli, V.; Parrinello, M. Accelerating the Calculation of Protein–Ligand Binding Free Energy and Residence Times Using Dynamically Optimized Collective Variables. J. Chem. Theory Comput. 2019, 15, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, C.; McClain, M.J.; Manjavacas, A.; Krauter, C.M.; Tian, S.; Berg, F.; Everitt, H.O.; Carter, E.A.; Nordlander, P.; et al. Aluminum Nanocrystals as a Plasmonic Photocatalyst for Hydrogen Dissociation. Nano Lett. 2016, 16, 1478–1484. [Google Scholar] [CrossRef]
- Swearer, D.F.; Robatjazi, H.; Martirez, J.M.P.; Zhang, M.; Zhou, L.; Carter, E.A.; Nordlander, P.; Halas, N.J. Plasmonic Photocatalysis of Nitrous Oxide into N2 and O2 Using Aluminum–Iridium Antenna–Reactor Nanoparticles. ACS Nano 2019, 13, 8076–8086. [Google Scholar] [CrossRef]
- Chen, Y.; Xin, X.; Zhang, N.; Xu, Y.J. Aluminum-Based Plasmonic Photocatalysis. Part. Part. Syst. Charact. 2017, 34, 1600357. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Lou, M.; Clark, B.D.; Lou, M.; Zhou, L.; Tian, S.; Jacobson, C.R.; Nordlander, P.; Halas, N.J. Morphology-Dependent Reactivity of a Plasmonic Photocatalyst. ACS Nano 2020, 14, 12054–12063. [Google Scholar] [CrossRef]
- Robatjazi, H.; Lou, M.; Clark, B.D.; Jacobson, C.R.; Swearer, D.F.; Nordlander, P.; Halas, N.J. Site-Selective Nanoreactor Deposition on Photocatalytic Al Nanocubes. Nano Lett. 2020, 20, 4550–4557. [Google Scholar] [CrossRef]
- Renard, D.; Tian, S.; Ahmadivand, A.; DeSantis, C.J.; Clark, B.D.; Nordlander, P.; Halas, N.J. Polydopamine-Stabilized Aluminum Nanocrystals: Aqueous Stability and Benzo[a]pyrene Detection. ACS Nano 2019, 13, 3117–3124. [Google Scholar] [CrossRef]
- Schwoebel, R.L.; Shipsey, E.J. Step Motion on Crystal Surfaces. J. Appl. Phys. 1966, 37, 3682–3686. [Google Scholar] [CrossRef]
- Mahmoud, S.; Carrez, P.; Landeiro Dos Reis, M.; Mousseau, N.; Cordier, P. Diffusion mechanism of bound Schottky defect in magnesium oxide. Phys. Rev. Mater. 2021, 5, 033609. [Google Scholar] [CrossRef]
- Zheng, J.; Ju, Z.; Zhang, B.; Nai, J.; Liu, T.; Liu, Y.; Xie, Q.; Zhang, W.; Wang, Y.; Tao, X. Lithium ion diffusion mechanism on the inorganic components of the solid–electrolyte interphase. J. Mater. Chem. A 2021, 9, 10251–10259. [Google Scholar] [CrossRef]
- Šimonová Baranyaiová, T.; Mészáros, R.; Sebechlebská, T.; Bujdák, J. Non-Arrhenius kinetics and slowed-diffusion mechanism of molecular aggregation of a rhodamine dye on colloidal particles. Phys. Chem. Chem. Phys. 2021, 23, 17177–17185. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Albe, K. Influence of Br−/S2− site-exchange on Li diffusion mechanism in Li6PS5Br: A computational study. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2021, 379, 20190458. [Google Scholar] [CrossRef] [PubMed]
- McCarty, K.F.; Nobel, J.A.; Bartelt, N.C. Vacancies in solids and the stability of surface morphology. Nature 2001, 412, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Cowern, N.E.B.; Simdyankin, S.; Ahn, C.; Bennett, N.S.; Goss, J.P.; Hartmann, J.M.; Pakfar, A.; Hamm, S.; Valentin, J.; Napolitani, E.; et al. Extended Point Defects in Crystalline Materials: Ge and Si. Phys. Rev. Lett. 2013, 110, 155501. [Google Scholar] [CrossRef] [Green Version]
- Südkamp, T.; Bracht, H. Self-diffusion in crystalline silicon: A single diffusion activation enthalpy down to 755 °C. Phys. Rev. B 2016, 94, 125208. [Google Scholar] [CrossRef]
- Ishikawa, R.; Mishra, R.; Lupini, A.R.; Findlay, S.D.; Taniguchi, T.; Pantelides, S.T.; Pennycook, S.J. Direct Observation of Dopant Atom Diffusion in a Bulk Semiconductor Crystal Enhanced by a Large Size Mismatch. Phys. Rev. Lett. 2014, 113, 155501. [Google Scholar] [CrossRef] [Green Version]
- Xiong, G.; Clark, J.N.; Nicklin, C.; Rawle, J.; Robinson, I.K. Atomic Diffusion within Individual Gold Nanocrystal. Sci. Rep. 2014, 4, 6765. [Google Scholar] [CrossRef] [Green Version]
- Windl, W.; Bunea, M.M.; Stumpf, R.; Dunham, S.T.; Masquelier, M.P. First-Principles Study of Boron Diffusion in Silicon. Phys. Rev. Lett. 1999, 83, 4345–4348. [Google Scholar] [CrossRef]
- Dolan, M.D.; McLennan, K.G.; Way, J.D. Diffusion of Atomic Hydrogen through V–Ni Alloy Membranes under Nondilute Conditions. J. Phys. Chem. C 2012, 116, 1512–1518. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Smidstrup, S.; Markussen, T.; Vancraeyveld, P.; Wellendorff, J.; Schneider, J.; Gunst, T.; Verstichel, B.; Stradi, D.; Khomyakov, P.A.; Vej-Hansen, U.G.; et al. QuantumATK: An integrated platform of electronic and atomic-scale modelling tools. J. Phys. Condens. Matter 2020, 32, 015901. [Google Scholar] [CrossRef]
- Winey, J.M.; Kubota, A.; Gupta, Y.M. A thermodynamic approach to determine accurate potentials for molecular dynamics simulations: Thermoelastic response of aluminum. Model. Simul. Mater. Sci. Eng. 2009, 17, 055004. [Google Scholar] [CrossRef]
- Chandrasekhar, S. Stochastic Problems in Physics and Astronomy. Rev. Mod. Phys. 1943, 15, 1–89. [Google Scholar] [CrossRef]
- van Gunsteren, W.; Berendsen, H. Algorithms for brownian dynamics. Mol. Phys. 1982, 45, 637–647. [Google Scholar] [CrossRef]
- Cooper, A. Precise lattice constsants of germanium, aluminum, gallium arsenide, uranium, sulphur, quartz and sapphire. Acta Crystallogr. 1962, 15, 578–582. [Google Scholar] [CrossRef] [Green Version]
- Finnis, M.W.; Heine, V. Theory of lattice contraction at aluminium surfaces. J. Phys. F Met. Phys. 1974, 4, L37–L41. [Google Scholar] [CrossRef]
- Ensing, B.; De Vivo, M.; Liu, Z.; Moore, P.; Klein, M.L. Metadynamics as a Tool for Exploring Free Energy Landscapes of Chemical Reactions. Accounts Chem. Res. 2006, 39, 73–81. [Google Scholar] [CrossRef]
- Kondati Natarajan, S.; Behler, J. Self-Diffusion of Surface Defects at Copper–Water Interfaces. J. Phys. Chem. C 2017, 121, 4368–4383. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mokkath, J.H.; Muhammed, M.M.; Chamkha, A.J. Free Energy Surfaces and Barriers for Vacancy Diffusion on Al(100), Al(110), Al(111) Reconstructed Surfaces. Nanomaterials 2022, 12, 76. https://doi.org/10.3390/nano12010076
Mokkath JH, Muhammed MM, Chamkha AJ. Free Energy Surfaces and Barriers for Vacancy Diffusion on Al(100), Al(110), Al(111) Reconstructed Surfaces. Nanomaterials. 2022; 12(1):76. https://doi.org/10.3390/nano12010076
Chicago/Turabian StyleMokkath, Junais Habeeb, Mufasila Mumthaz Muhammed, and Ali J. Chamkha. 2022. "Free Energy Surfaces and Barriers for Vacancy Diffusion on Al(100), Al(110), Al(111) Reconstructed Surfaces" Nanomaterials 12, no. 1: 76. https://doi.org/10.3390/nano12010076