
Behaviour of PMMA Resin Composites Incorporated with Nanoparticles or Fibre following Prolonged Water Storage

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Specimen Preparation
2.2.2. Silanisation of Nano-ZrO2 and Nano-TiO2
2.2.3. Blending Filler with PMMA/MMA
2.2.4. Water Sorption and Solubility Measurement
2.2.5. Measurement of Hygroscopic Dimensional Changes
2.3. Statistical Analysis
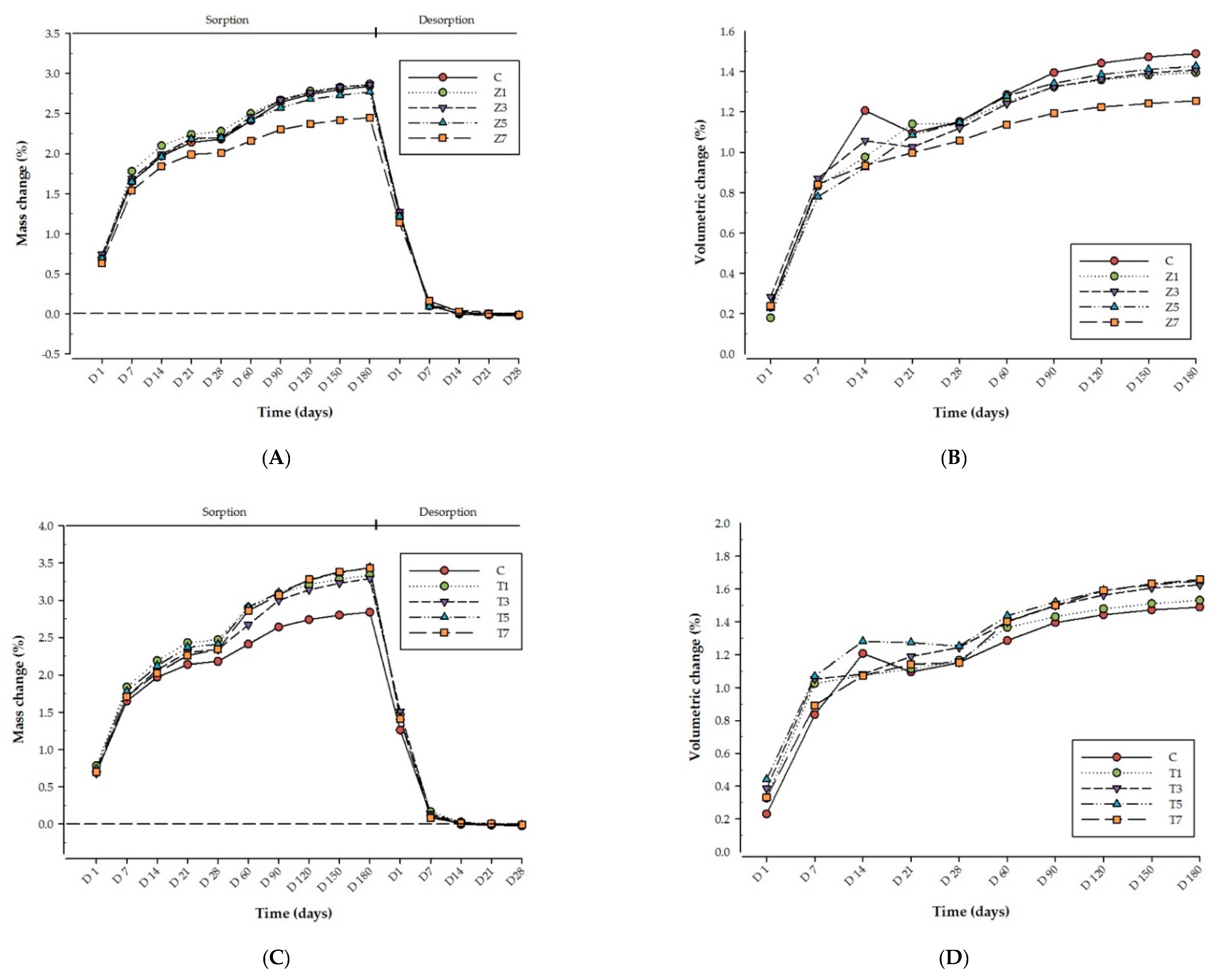
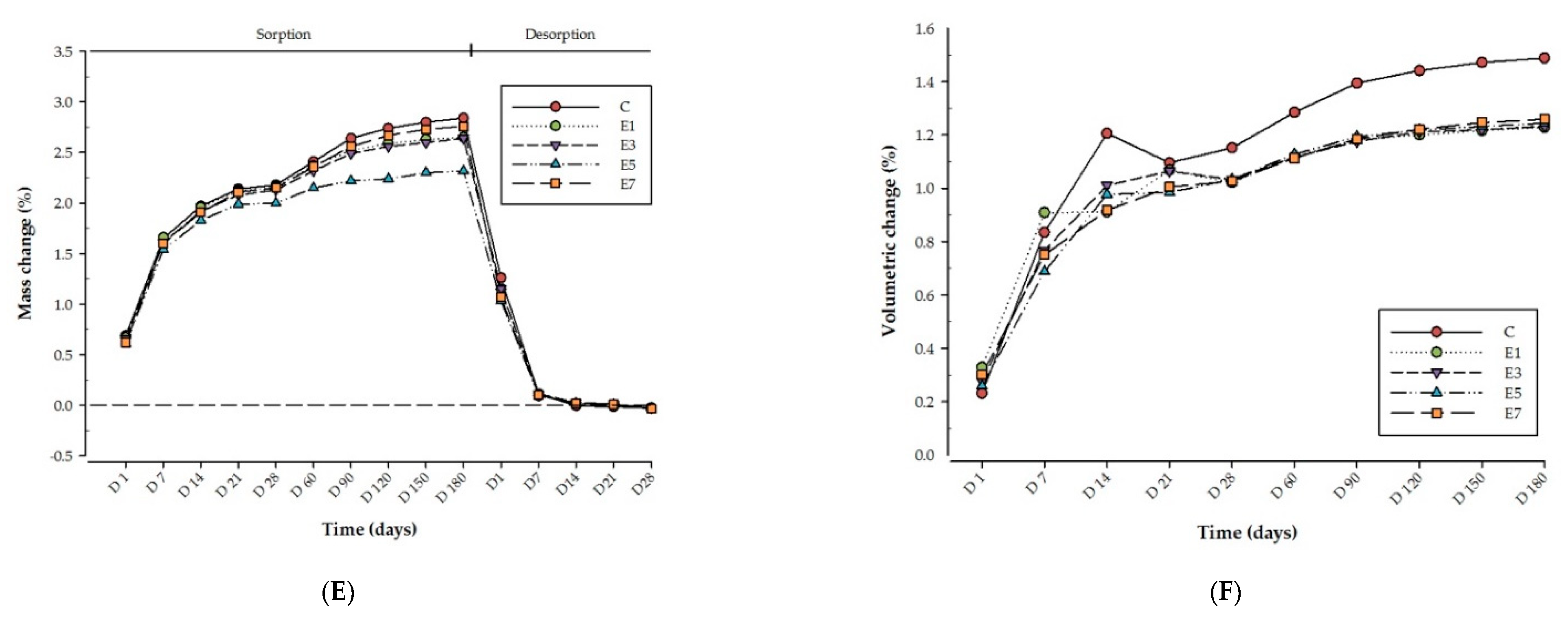
3. Results
4. Discussion
Clinical Significance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parvizi, A.; Lindquist, T.; Schneider, R.; Williamson, D.; Boyer, D.; Dawson, D.V. Comparison of the Dimensional Accuracy of Injection-Molded Denture Base Materials to that of Conventional Pressure-Pack Acrylic Resin. J. Prosthodont. 2004, 13, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Demir, H.; Dogan, O.M.; Dogan, A. Water sorption of denture base resin reinforced with different fibres. Mater. Res. Innov. 2010, 14, 332–337. [Google Scholar] [CrossRef]
- Wei, Y.-J.; Silikas, N.; Zhang, Z.-T.; Watts, D. Hygroscopic dimensional changes of self-adhering and new resin-matrix composites during water sorption/desorption cycles. Dent. Mater. 2011, 27, 259–266. [Google Scholar] [CrossRef]
- Polat, T.N.; Karacaer, Ö.; Tezvergil, A.; Lassila, L.V.J.; Vallittu, P.K. Water Sorption, Solubility and Dimensional Changes of Denture Base Polymers Reinforced with Short Glass Fibers. J. Biomater. Appl. 2003, 17, 321–335. [Google Scholar] [CrossRef]
- Sasaki, H.; Hamanaka, I.; Takahashi, Y.; Kawaguchi, T. Effect of long-term water immersion or thermal shock on mechanical properties of high-impact acrylic denture base resins. Dent. Mater. J. 2016, 35, 204–209. [Google Scholar] [CrossRef] [Green Version]
- Asar, N.V.; Albayrak, H.; Korkmaz, T.; Turkyilmaz, I. Influence of various metal oxides on mechanical and physical properties of heat-cured polymethyl methacrylate denture base resins. J. Adv. Prosthodont. 2013, 5, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, V.M.; Vallittu, P.K. Water sorption and solubility of glass fiber-reinforced denture polymethyl methacrylate resin. J. Prosthet. Dent. 1997, 77, 531–534. [Google Scholar] [CrossRef]
- Anttila, E.J.; Krintila, O.H.; Laurila, T.K.; Lassila, L.V.; Vallittu, P.K.; Hernberg, R.G. Evaluation of polymerization shrinkage and hydroscopic expansion of fiber-reinforced biocomposites using optical fiber Bragg grating sensors. Dent. Mater. 2008, 24, 1720–1727. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Rosenbauer, E.-U. Residual methyl methacrylate monomer, water sorption, and water solubility of hypoallergenic denture base materials. J. Prosthet. Dent. 2004, 92, 72–78. [Google Scholar] [CrossRef]
- Wei, Y.-J.; Silikas, N.; Zhang, Z.-T.; Watts, D. The relationship between cyclic hygroscopic dimensional changes and water sorption/desorption of self-adhering and new resin-matrix composites. Dent. Mater. 2013, 29, e218–e226. [Google Scholar] [CrossRef]
- Sideridou, I.; Tserki, V.; Papanastasiou, G.J.B. Study of water sorption, solubility and modulus of elasticity of light-cured dimethacrylate-based dental resins. Biomaterials 2003, 24, 655–665. [Google Scholar] [CrossRef]
- Martin, N.; Jedynakiewicz, N.M.; Fisher, A.C. Hygroscopic expansion and solubility of composite restoratives. Dent. Mater. 2003, 19, 77–86. [Google Scholar] [CrossRef]
- Cucci, A.L.; Vergani, C.; Giampaolo, E.T.; Afonso, M.C.S.F. Water sorption, solubility, and bond strength of two autopolymerizing acrylic resins and one heat-polymerizing acrylic resin. J. Prosthet. Dent. 1998, 80, 434–438. [Google Scholar] [CrossRef]
- Dhir, G.; Berzins, D.W.; Dhuru, V.B.; Periathamby, A.R.; Dentino, A. Physical Properties of Denture Base Resins Potentially Resistant to Candida Adhesion. J. Prosthodont. 2007, 16, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuna, S.H.; Keyf, F.; Gumus, H.O.; Uzun, C. The Evaluation of Water Sorption/Solubility on Various Acrylic Resins. Eur. J. Dent. 2008, 2, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Zidan, S.; Silikas, N.; Haider, J.; Yates, J. Long-Term Sorption and Solubility of Zirconia-Impregnated PMMA Nanocomposite in Water and Artificial Saliva. Materials 2020, 13, 3732. [Google Scholar] [CrossRef]
- Zankuli, M.; Devlin, H.; Silikas, N. Water sorption and solubility of core build-up materials. Dent. Mater. 2014, 30, e324–e329. [Google Scholar] [CrossRef]
- Ilie, N.; Hilton, T.; Heintze, S.; Hickel, R.; Watts, D.; Silikas, N.; Stansbury, J.; Cadenaro, M.; Ferracane, J. Academy of Dental Materials guidance—resin composites: Part I—Mechanical properties. Dent. Mater. 2017, 33, 880–894. [Google Scholar] [CrossRef]
- Ruttermann, S.; Kruger, S.; Raab, W.H.-M.; Janda, R. Polymerization shrinkage and hygroscopic expansion of contemporary posterior resin-based filling materials—A comparative study. J. Dent. 2007, 35, 806–813. [Google Scholar] [CrossRef]
- Vallittu, P.K. Effect of 180-week water storage on the flexural properties of E-glass and silica fiber acrylic resin composite. Int. J. Prosthodont. 2001, 13, 13. [Google Scholar]
- Miettinen, V.M.; Narva, K.K.; Vallittu, P.K. Water sorption, solubility and effect of post-curing of glass fibre reinforced polymers. Biomaterials 1999, 20, 1187–1194. [Google Scholar] [CrossRef]
- Jagger, R.G. Effect of the curing cycle on some properties of a polymethylmethacrylate denture base material. J. Oral Rehabil. 1978, 5, 151–157. [Google Scholar] [CrossRef]
- Hamanaka, I.; Iwamoto, M.; Lassila, L.; Vallittu, P.; Shimizu, H.; Takahashi, Y. Influence of water sorption on mechanical properties of injection-molded thermoplastic denture base resins. Acta Odontol. Scand. 2014, 72, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.M.; Almeida, G.S.; Poskus, L.T.; Guimaraes, J.G.A. Relationship between the degree of conversion, solubility and salivary sorption of a hybrid and a nanofilled resin composite. J. Appl. Oral Sci. 2008, 16, 161–166. [Google Scholar] [CrossRef] [Green Version]
- ISO. ISO:20795-1 Dentistry—Denture Base Polymers; ISO: Geneva, Switzerland, 2008. [Google Scholar]
- Yang, A.; Zhao, D.; Wu, Y.; Xu, C. Effect of polyimide addition on mechanical properties of PMMA-based denture material. Dent. Mater. J. 2017, 36, 560–565. [Google Scholar] [CrossRef] [Green Version]
- Gad, M.M.; Al-Thobity, A.M.; Rahoma, A.; Abualsaud, R.; Al-Harbi, F.A.; Akhtar, S. Reinforcement of PMMA Denture Base Material with a Mixture of ZrO2 Nanoparticles and Glass Fibers. Int. J. Dent. 2019, 2019, 2489393. [Google Scholar] [CrossRef] [Green Version]
- Alhotan, A.; Yates, J.; Zidan, S.; Haider, J.; Silikas, N. Flexural Strength and Hardness of Filler-Reinforced PMMA Targeted for Denture Base Application. Materials 2021, 14, 2659. [Google Scholar] [CrossRef] [PubMed]
- Alhotan, A.; Yates, J.; Zidan, S.; Haider, J.; Silikas, N. Assessing Fracture Toughness and Impact Strength of PMMA Reinforced with Nano-Particles and Fibre as Advanced Denture Base Materials. Materials 2021, 14, 4127. [Google Scholar] [CrossRef]
- Gad, M.M.; Abualsaud, R.; Rahoma, A.; Al-Thobity, A.M.; Al-Abidi, K.S.; Akhtar, S. Effect of zirconium oxide nanoparticles addition on the optical and tensile properties of polymethyl methacrylate denture base material. Int. J. Nanomed. 2018, 13, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leao, R.D.S.; de Moraes, S.L.D.; Gomes, J.M.D.L.; Lemos, C.A.A.; Casado, B.G.D.S.; Vasconcelos, B.C.D.E.; Pellizzer, E.P. Influence of addition of zirconia on PMMA: A systematic review. Mater. Sci. Eng. C 2020, 106, 110292. [Google Scholar] [CrossRef]
- Ohkubo, C.; Hanatani, S.; Hosoi, T. Present status of titanium removable dentures–a review of the literature. J. Oral Rehabil. 2008, 35, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Matinlinna, J.P. E-Glass Fiber Reinforced Composites in Dental Applications. Silicon 2012, 4, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Alshabib, A.; Algamaiah, H.; Silikas, N.; Watts, D.C. Material behavior of resin composites with and without fibers after extended water storage. Dent. Mater. J. 2021, 40, 557–565. [Google Scholar] [CrossRef]
- Alshali, R.Z.; Salim, N.A.; Satterthwaite, J.D.; Silikas, N. Long-term sorption and solubility of bulk-fill and conventional resin-composites in water and artificial saliva. J. Dent. 2015, 43, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Ferracane, J.L. Hygroscopic and hydrolytic effects in dental polymer networks. Dent. Mater. 2006, 22, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, A.-M.; Dyer, S.R.; Lassila, L.V.; Vallittu, P.K. Effect of rigid rod polymer filler on mechanical properties of poly-methyl methacrylate denture base material. Dent. Mater. 2008, 24, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Durkan, R.K.; Ozdemir, T.; Pamir, A.D.; Usanmaz, A. Water absorption of two different denture base resins reinforced with dental fiber systems. J. Appl. Polym. Sci. 2010, 117, 1750–1753. [Google Scholar] [CrossRef]
- Lung, C.Y.K.; Sarfraz, Z.; Habib, A.; Khan, A.S.; Matinlinna, J.P. Effect of silanization of hydroxyapatite fillers on physical and mechanical properties of a bis-GMA based resin composite. J. Mech. Behav. Biomed. Mater. 2016, 54, 283–294. [Google Scholar] [CrossRef]
- Wei, Y.-J.; Silikas, N.; Zhang, Z.-T.; Watts, D.C. Diffusion and concurrent solubility of self-adhering and new resin–matrix composites during water sorption/desorption cycles. Dent. Mater. 2011, 27, 197–205. [Google Scholar] [CrossRef]
- Chladek, G.; Pakiela, K.; Pakiela, W.; Zmudzki, J.; Adamiak, M.; Krawczyk, C. Effect of Antibacterial Silver-Releasing Filler on the Physicochemical Properties of Poly(Methyl Methacrylate) Denture Base Material. Materials 2019, 12, 4146. [Google Scholar] [CrossRef] [Green Version]
- Zidan, S.; Silikas, N.; Alhotan, A.; Haider, J.; Yates, J. Investigating the Mechanical Properties of ZrO2-Impregnated PMMA Nanocomposite for Denture-Based Applications. Materials 2019, 12, 1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ergun, G.; Sahin, Z.; Ataol, A.S. The effects of adding various ratios of zirconium oxide nanoparticles to poly(methyl methacrylate) on physical and mechanical properties. J. Oral Sci. 2018, 60, 304–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundie, F.; Azhari, C.; Ahmad, Z.A. Effect of nano- and micro-alumina fillers on some properties of poly(methyl methacrylate) denture base composites. J. Serb. Chem. Soc. 2018, 83, 75–91. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, V.M.; Vallittu, P.K. Release of residual methyl methacrylate into water from glass fibre-poly(methyl methacrylate) composite used in dentures. Biomaterials 1997, 18, 181–185. [Google Scholar] [CrossRef]
- Kondo, Y.; Takagaki, T.; Okuda, M.; Ikeda, M.; Kadoma, Y.; Yamauchi, J.; Okada, K.; Sadr, A.; Nikaido, T.; Tagami, J. Effect of PMMA filler particles addition on the physical properties of resin composite. Dent. Mater. J. 2010, 29, 596–601. [Google Scholar] [CrossRef] [Green Version]
- Garcia, L.d.F.R.; Roselino, L.d.M.R.; Mundim, F.M.; Pires-de-Souza, F.d.C.P.; Consani, S. Influence of artificial accelerated aging on dimensional stability of acrylic resins submitted to different storage protocols. J. Prosthodont. Implant. Esthet. Reconstr. Dent. 2010, 19, 432–437. [Google Scholar] [CrossRef]
- Wong, D.M.; Cheng, L.Y.; Chow, T.; Clark, R.K. Effect of processing method on the dimensional accuracy and water sorption of acrylic resin dentures. J. Prosthet. Dent. 1999, 81, 300–304. [Google Scholar] [CrossRef]
- Chow, T.; Cheng, Y.; Ladizesky, N. Polyethylene fibre reinforced poly(methylmethacrylate)—water sorption and dimensional changes during immersion. J. Dent. 1993, 21, 367–372. [Google Scholar] [CrossRef]
- Ladizesky, N.H.; Chow, T.W.; Cheng, Y.Y. Denture base reinforcement using woven polyethylene fiber. Int. J. Prosthodont. 1994, 7, 307–314. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Materials Group | Wsp (µg/mm3) | Ms% | Wsl (µg/mm3) | Md% | Hygroscopic Expansion (%) | |
|---|---|---|---|---|---|---|
| Control | C | 31.6 (4.9) ac | 2.84 (0.48) ac | 0.27 (0.17) abc | −0.024 (0.016) abc | 1.49 (0.18) abc |
| ZrO2 | Z%1.5 | 31.1 (6.8) a | 2.87 (0.68) a | 0.15 (0.13) a | −0.014 (0.012) a | 1.38 (0.29) a |
| Z%3 | 32.0 (5.3) a | 2.84 (0.51) a | 0.13 (0.48) a | −0.011 (0.004) a | 1.41 (0.20) a | |
| Z%5 | 30.9 (5.8) a | 2.73 (0.55) a | 0.14 (0.17) a | −0.013 (0.015) a | 1.43 (0.26) a | |
| Z%7 | 27.9 (3.6) a | 2.44 (0.32) a | 0.13 (0.07) a | −0.012 (0.006) a | 1.25 (0.16) a | |
| TiO2 | T%1.5 | 35.6 (0.7) b | 3.33 (0.08) b | 0.22 (0.20) b | −0.021 (0.020) b | 1.53 (0.33) b |
| T%3 | 35.9 (0.7) b | 3.29 (0.09) b | 0.16 (0.17) b | −0.015 (0.017) b | 1.62 (0.37) b | |
| T%5 | 37.8 (0.5) b | 3.43 (0.08) b | 0.12 (0.15) b | −0.011 (0.014) b | 1.65 (0.25) b | |
| T%7 | 38.5 (0.7) b | 3.44 (0.07) b | 0.11 (0.06) b | −0.010 (0.005) b | 1.66 (0.41) b | |
| E-glass fibre | E%1.5 | 29.6 (6.2) c | 2.65 (0.64) c | 0.26 (0.32) c | −0.023 (0.027) c | 1.22 (0.11) c |
| E%3 | 30.0 (6.4) c | 2.64 (0.68) c | 0.30 (0.06) c | −0.026 (0.005) c | 1.23 (0.08) c | |
| E%5 | 27.7 (3.3) c | 2.32 (0.26) c | 0.33 (0.13) c | −0.028 (0.011) c | 1.24 (0.12) c | |
| E%7 | 31.9 (5.0) c | 2.76 (0.49) c | 0.43 (0.11) c | −0.037 (0.009) c | 1.26 (0.11) c | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhotan, A.; Yates, J.; Zidan, S.; Haider, J.; Jurado, C.A.; Silikas, N. Behaviour of PMMA Resin Composites Incorporated with Nanoparticles or Fibre following Prolonged Water Storage. Nanomaterials 2021, 11, 3453. https://doi.org/10.3390/nano11123453
Alhotan A, Yates J, Zidan S, Haider J, Jurado CA, Silikas N. Behaviour of PMMA Resin Composites Incorporated with Nanoparticles or Fibre following Prolonged Water Storage. Nanomaterials. 2021; 11(12):3453. https://doi.org/10.3390/nano11123453
Chicago/Turabian StyleAlhotan, Abdulaziz, Julian Yates, Saleh Zidan, Julfikar Haider, Carlos Alberto Jurado, and Nikolaos Silikas. 2021. "Behaviour of PMMA Resin Composites Incorporated with Nanoparticles or Fibre following Prolonged Water Storage" Nanomaterials 11, no. 12: 3453. https://doi.org/10.3390/nano11123453