Detonation Nanodiamonds: A Comparison Study by Photoacoustic, Diffuse Reflectance, and Attenuated Total Reflection FTIR Spectroscopies


Abstract
:1. Introduction
2. Materials and Methods
2.1. Nanodiamonds
2.2. Instrumentation
2.2.1. FTIR–PAS
2.2.2. ATR–FTIR
2.2.3. DRIFT
2.3. Data Handling
2.4. Procedures
Reproducibility
3. Results
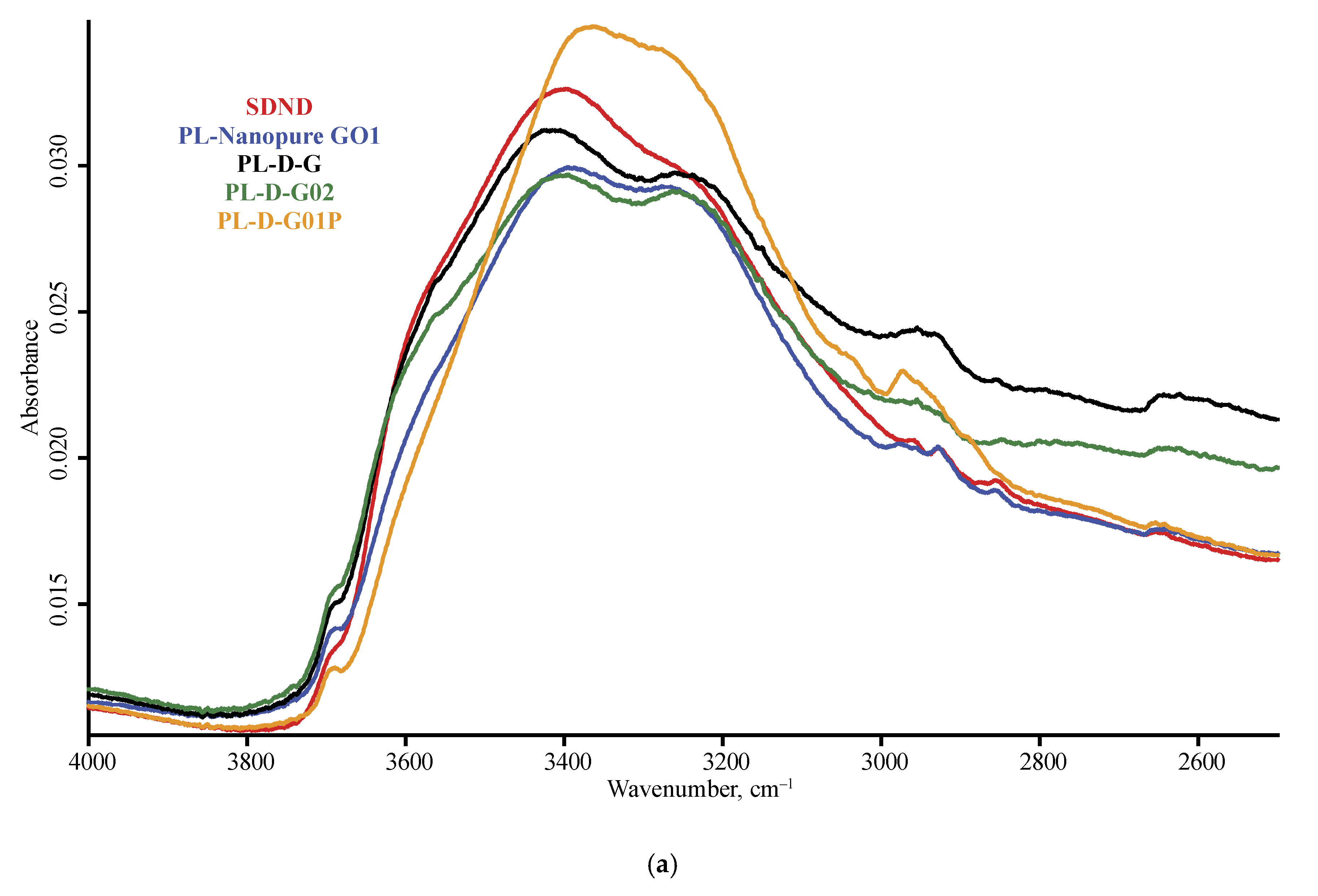
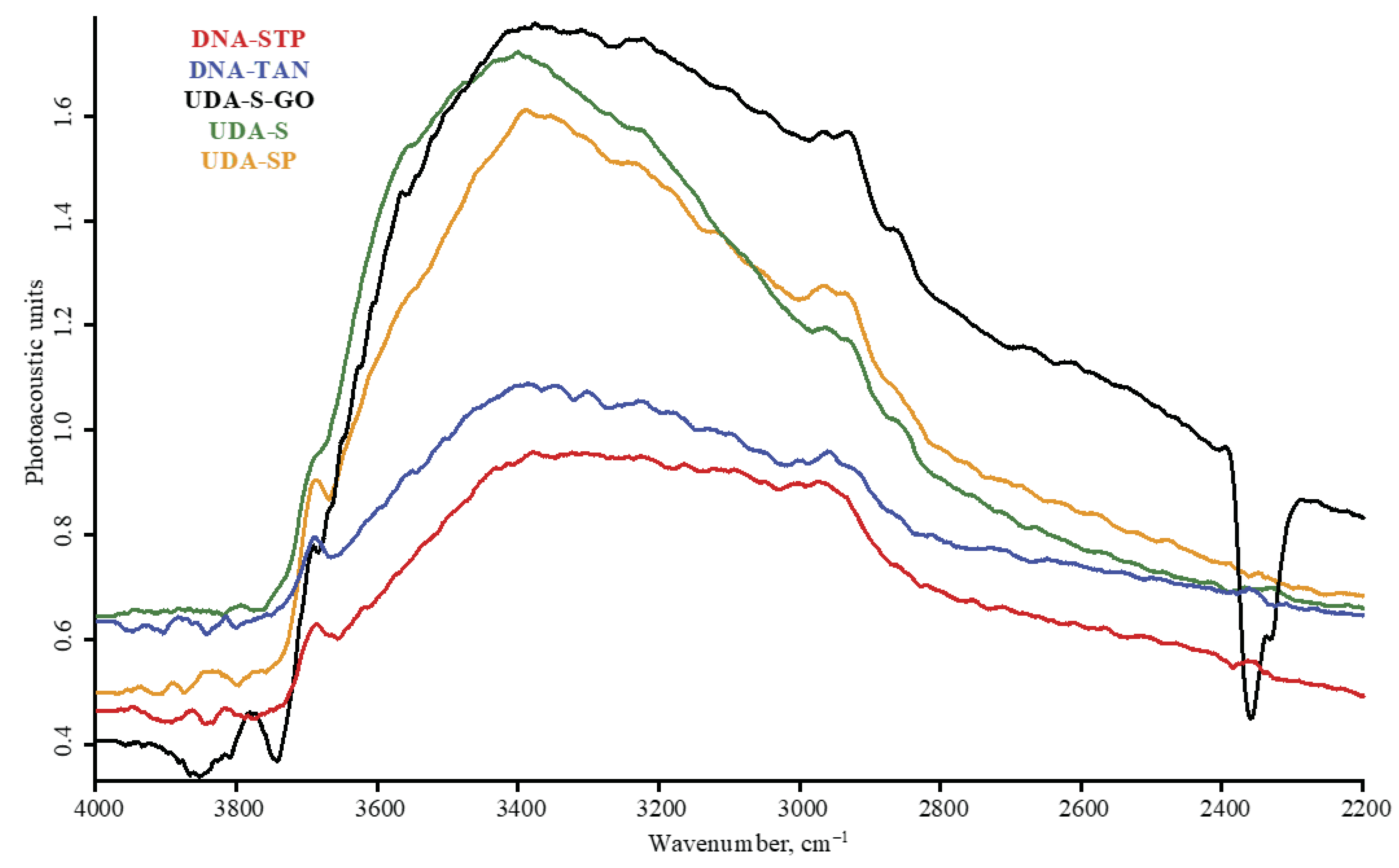
3.1. Band Assignment
3.2. Signal-Gathering Depth and FTIR–PAS Modulation Frequency Comparison
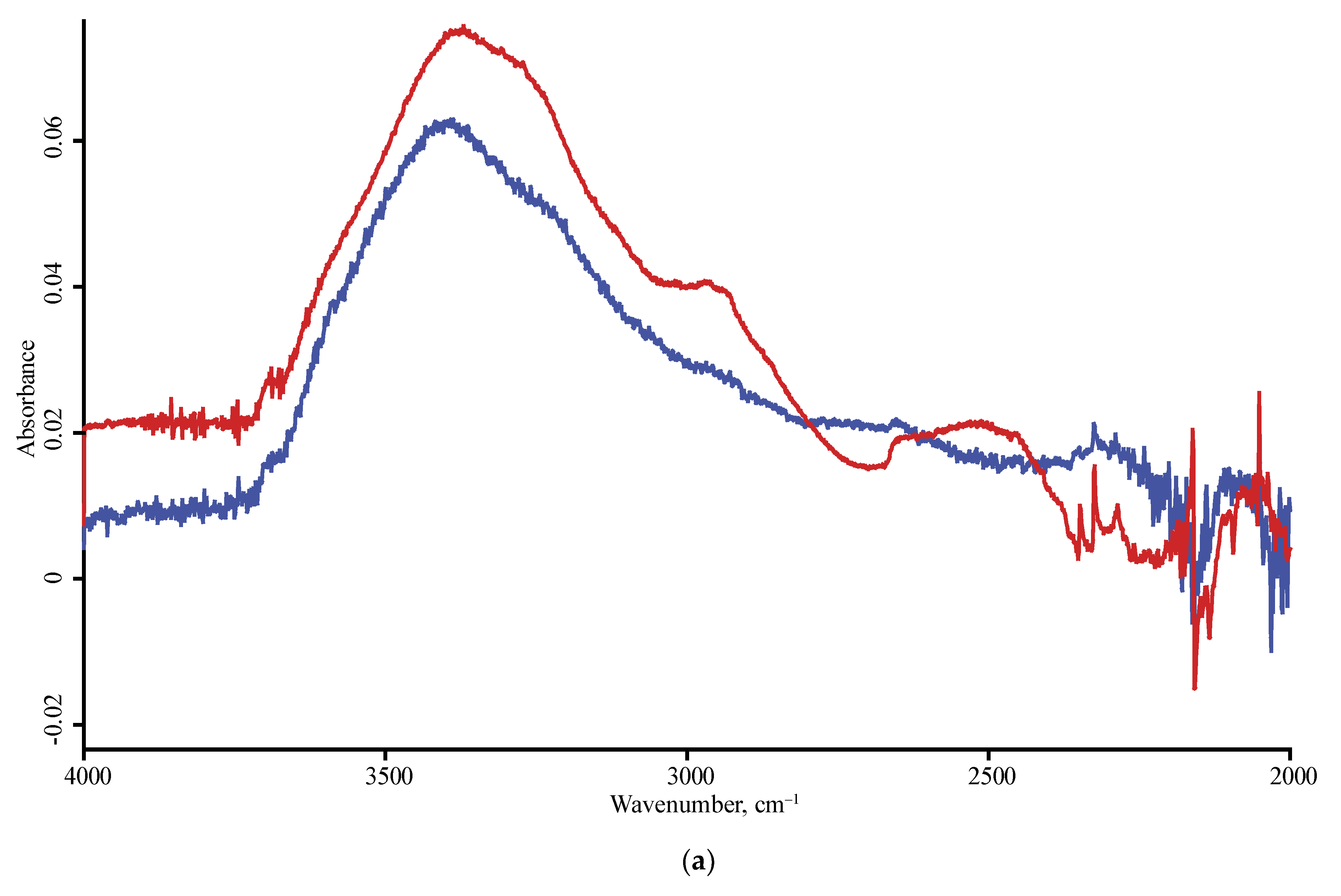
3.3. Band Reproducibility
4. Discussion
4.1. DRIFT
4.2. ATR–FTIR
4.3. FTIR–PAS
4.4. ND Brand Features
4.5. Modality Comparison
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A








References
- Buchatskaya, Y.; Romanchuk, A.Y.; Yakovlev, R.; Shiryaev, A.; Kulakova, I.; Kalmykov, S. Sorption of actinides onto nanodiamonds. Radiochim. Acta 2015, 103, 205–211. [Google Scholar] [CrossRef]
- Stehlik, S.; Varga, M.; Ledinsky, M.; Jirasek, V.; Artemenko, A.; Kozak, H.; Ondic, L.; Skakalova, V.; Argentero, G.; Pennycook, T.; et al. Size and Purity Control of HPHT Nanodiamonds down to 1 nm. J. Phys. Chem. C 2015, 119, 27708–27720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peristyy, A.; Koreshkova, A.; Paull, B.; Nesterenko, P.N. Ion-exchange properties of High Pressure High Temperature synthetic diamond. Diam. Relat. Mater. 2017, 75, 131–139. [Google Scholar] [CrossRef]
- Peristyy, A.; Paull, B.; Nesterenko, P.N. Ion-exchange properties of microdispersed sintered detonation nanodiamond. Adsorption 2016, 22, 371–383. [Google Scholar] [CrossRef]
- Volkov, D.S.; Krivoshein, P.K.; Mikheev, I.V.; Proskurnin, M.A. Pristine detonation nanodiamonds as regenerable adsorbents for metal cations. Diam. Relat. Mater. 2020, 110, 108121. [Google Scholar] [CrossRef]
- Dolmatov, V.Y. Detonation-synthesis nanodiamonds: Synthesis, structure, properties and applications. Russ. Chem. Rev. 2007, 76, 339–360. [Google Scholar] [CrossRef]
- Chernysheva, M.G.; Popov, A.G.; Tashlitsky, V.N.; Badun, G.A. Cationic surfactant coating nanodiamonds: Adsorption and peculiarities. Colloids Surfaces A Physicochem. Eng. Asp. 2019, 565, 25–29. [Google Scholar] [CrossRef]
- Shenderova, O.A.; Gruen, D.M. Ultrananocrystalline Diamond, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Zhang, K.; Zhao, Q.; Qin, S.; Fu, Y.; Liu, R.; Zhi, J.; Shan, C.-X. Nanodiamonds conjugated upconversion nanoparticles for bio-imaging and drug delivery. J. Colloid Interface Sci. 2019, 537, 316–324. [Google Scholar] [CrossRef]
- Qin, S.-R.; Zhao, Q.; Cheng, Z.-G.; Zhang, D.-X.; Zhang, K.-K.; Su, L.-X.; Fan, H.-J.; Wang, Y.-H.; Shan, C.-X. Rare earth-functionalized nanodiamonds for dual-modal imaging and drug delivery. Diam. Relat. Mater. 2019, 91, 173–182. [Google Scholar] [CrossRef]
- Mochalin, V.N.; Pentecost, A.; Li, X.-M.; Neitzel, I.; Nelson, M.; Wei, C.; He, T.; Guo, F.; Gogotsi, Y. Adsorption of Drugs on Nanodiamond: Toward Development of a Drug Delivery Platform. Mol. Pharm. 2013, 10, 3728–3735. [Google Scholar] [CrossRef]
- Ţucureanu, V.; Matei, A.; Avram, A.M. FTIR Spectroscopy for Carbon Family Study. Crit. Rev. Anal. Chem. 2016, 46, 502–520. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ding, Y.; Zhang, B.; Huang, R.; Su, D.S. New insights into the oxidative dehydrogenation of propane on borate-modified nanodiamond. Chem. Commun. 2015, 51, 9145–9148. [Google Scholar] [CrossRef] [PubMed]
- Remes, Z.; Kozak, H.; Rezek, B.; Ukraintsev, E.; Babchenko, O.; Kromka, A.; Girard, H.A.; Arnault, J.-C.; Bergonzo, P. Diamond-coated ATR prism for infrared absorption spectroscopy of surface-modified diamond nanoparticles. Appl. Surf. Sci. 2013, 270, 411–417. [Google Scholar] [CrossRef]
- Stehlik, S.; Glatzel, T.; Pichot, V.; Pawlak, R.; Meyer, E.; Spitzer, D.; Rezek, B. Water interaction with hydrogenated and oxidized detonation nanodiamonds—Microscopic and spectroscopic analyses. Diam. Relat. Mater. 2016, 63, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Petit, T.; Puskar, L. FTIR spectroscopy of nanodiamonds: Methods and interpretation. Diam. Relat. Mater. 2018, 89, 52–66. [Google Scholar] [CrossRef]
- Mayerhöfer, T.G.; Mutschke, H.; Popp, J. Employing Theories Far beyond Their Limits-The Case of the (Boguer-) Beer-Lambert Law. ChemPhysChem 2016, 17, 1948–1955. [Google Scholar] [CrossRef]
- Zhang, J.; Su, D.S.; Blume, R.; Schlogl, R.; Wang, R.; Yang, X.; Gajovic, A. Surface chemistry and catalytic reactivity of a nanodiamond in the steam-free dehydrogenation of ethylbenzene. Angew. Chem. 2010, 122, 8822–8826. [Google Scholar] [CrossRef]
- Beyler-Çiğil, A.; Çakmakçı, E.; Vezir Kahraman, M. Thermal properties of phosphorylated nanodiamond reinforced polyimides. Polym. Compos. 2016, 37, 2285–2292. [Google Scholar] [CrossRef]
- Girard, H.A.; Perruchas, S.; Gesset, C.; Chaigneau, M.; Vieille, L.; Arnault, J.-C.; Bergonzo, P.; Boilot, J.-P.; Gacoin, T. Electrostatic Grafting of Diamond Nanoparticles: A Versatile Route to Nanocrystalline Diamond Thin Films. ACS Appl. Mater. Interfaces 2009, 1, 2738–2746. [Google Scholar] [CrossRef]
- Frosch, T.; Chan, K.L.A.; Wong, H.C.; Cabral, J.T.; Kazarian, S.G. Nondestructive Three-Dimensional Analysis of Layered Polymer Structures with Chemical Imaging. Langmuir 2010, 26, 19027–19032. [Google Scholar] [CrossRef]
- Inel, G.A.; Ungureau, E.-M.; Varley, T.S.; Hirani, M.; Holt, K.B. Solvent–surface interactions between nanodiamond and ethanol studied with in situ infrared spectroscopy. Diam. Relat. Mater. 2016, 61, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Xing, Z.; Tian, K.; Du, C.; Li, C.; Zhou, J.; Chen, Z. Agricultural soil characterization by FTIR spectroscopy at micrometer scales: Depth profiling by photoacoustic spectroscopy. Geoderma 2019, 335, 94–103. [Google Scholar] [CrossRef]
- Bauer, A.; Hertzberg, O.; Küderle, A.; Strobel, D.; Pleitez, M.A.; Mäntele, W. IR-spectroscopy of skin in vivo: Optimal skin sites and properties for non-invasive glucose measurement by photoacoustic and photothermal spectroscopy. J. Biophotonics 2018, 11, e201600261. [Google Scholar] [CrossRef] [PubMed]
- Brangule, A.; Skadiņš, I.; Reinis, A.; Gross, K.A.; Kroča, J. In Vitro Characterization Perspectives Using Fourier Transform Infrared Photoacoustic Spectroscopy (FTIR-PAS) and Diffuse Reflectance Infrared Spectroscopy (DRIFT). Key Eng. Mater. 2017, 758, 273–277. [Google Scholar] [CrossRef]
- Kizil, R.; Irudayaraj, J. Fourier Transform Infrared Photoacoustic Spectroscopy (FTIR-PAS). In Encyclopedia of Biophysics; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2013; pp. 840–844. [Google Scholar]
- Michaelian, K.H.; Wen, Q. Photoacoustic infrared spectroscopy of solids. J. Phys. Conf. Ser. 2010, 214, 012004. [Google Scholar] [CrossRef]
- Ando, T.; Inoue, S.; Ishii, M.; Kamo, M.; Sato, Y.; Yamada, O.; Nakano, T. Fourier-transform infrared photoacoustic studies of hydrogenated diamond surfaces. J. Chem. Soc. Faraday Trans. 1993, 89, 749. [Google Scholar] [CrossRef]
- Pasieczna-Patkowska, S.; Madej, J. Comparison of photoacoustic, diffuse reflectance, attenuated total reflectance and transmission infrared spectroscopy for the study of biochars. Pol. J. Chem. Technol. 2018, 20, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Krivoshein, P.K.; Volkov, D.S.; Rogova, O.B.; Proskurnin, M.A. FTIR photoacoustic spectroscopy for identification and assessment of soil components: Chernozems and their size fractions. Photoacoustics 2020, 18, 100162. [Google Scholar] [CrossRef]
- Du, C.; Zhou, J. Application of Infrared Photoacoustic Spectroscopy in Soil Analysis. Appl. Spectrosc. Rev. 2011, 46, 405–422. [Google Scholar] [CrossRef]
- Michaelian, K.H.; Photoacoustic, I.R. Spectroscopy: Instrumentation, Applications and Data Analysis; 2nd Revised and Enlarged Edition; Wiley-VCH: Weinheim, Germany, 2010; p. 402. [Google Scholar]
- Tokmakoff, A.; Banholzer, W.F.; Fayer, M.D. Thermal diffusivity measurements of natural and isotopically enriched diamond by picosecond infrared transient grating experiments. Appl. Phys. A 1993, 56, 87–90. [Google Scholar] [CrossRef]
- Kronenberg, A.K. Chapter 4. Hydrogen Speciation and Chemical Weakening of Quartz. Silica 1994, 123–176. [Google Scholar] [CrossRef]
- Spitsyn, B.; Davidson, J.; Gradoboev, M.; Galushko, T.; Serebryakova, N.; Karpukhina, T.; Kulakova, I.; Novikova, N.N. Inroad to modification of detonation nanodiamond. Diam. Relat. Mater. 2006, 15, 296–299. [Google Scholar] [CrossRef]
- Calderón, F.J.; Mikha, M.M.; Vigil, M.F.; Nielsen, D.C.; Benjamin, J.G.; Reeves, J.B. Diffuse-Reflectance Mid-infrared Spectral Properties of Soils under Alternative Crop Rotations in a Semi-arid Climate. Commun. Soil Sci. Plant Anal. 2011, 42, 2143–2159. [Google Scholar] [CrossRef]
- Hadzi, D.; Pintar, M. The OH in-plane deformation and the C-O stretching frequencies in monomeric carboxylic acids and their association shifts. Spectrochim. Acta 1958, 12, 162–168. [Google Scholar] [CrossRef]
- Asselin, M.; Sandorfy, C. Anharmonicity and Hydrogen Bonding. The in-plane OH Bending and its Combination with the OH Stretching Vibration. Can. J. Chem. 1971, 49, 1539–1544. [Google Scholar] [CrossRef] [Green Version]
- Hens, S.C.; Cunningham, G.; Tyler, T.; Moseenkov, S.; Kuznetsov, V.; Shenderova, O. Nanodiamond bioconjugate probes and their collection by electrophoresis. Diam. Relat. Mater. 2008, 17, 1858–1866. [Google Scholar] [CrossRef]
- Colthup, N.B.; Daly, L.H.; Wiberley, S.E. Introduction to Infrared and Raman Spectroscopy, 3rd ed.; Academic Press: New York, NY, USA, 1990; pp. 27–33. [Google Scholar]
- Mitev, D.; Dimitrova, R.; Spassova, M.; Minchev, C.; Stavrev, S. Surface peculiarities of detonation nanodiamonds in dependence of fabrication and purification methods. Diam. Relat. Mater. 2007, 16, 776–780. [Google Scholar] [CrossRef]
- Jiang, T.; Xu, K. FTIR study of ultradispersed diamond powder synthesied by explosive detonation. Carbon 1995, 33, 1663–1671. [Google Scholar] [CrossRef]
- Korobov, M.V.; Volkov, D.S.; Avramenko, N.V.; Belyaeva, L.A.; Semenyuk, P.I.; Proskurnin, M.A. Improving the dispersity of detonation nanodiamond: Differential scanning calorimetry as a new method of controlling the aggregation state of nanodiamond powders. Nanoscale 2013, 5, 1529–1536. [Google Scholar] [CrossRef]
- Tu, J.-S.; Perevedentseva, E.; Chung, P.-H.; Cheng, C.-L. Size-dependent surface CO stretching frequency investigations on nanodiamond particles. J. Chem. Phys. 2006, 125, 174713. [Google Scholar] [CrossRef]
- Volkov, D.S.; Proskurnin, M.A.; Korobov, M.V. Elemental analysis of nanodiamonds by inductively-coupled plasma atomic emission spectroscopy. Carbon 2014, 74, 1–13. [Google Scholar] [CrossRef]
- Volkov, D.S.; Proskurnin, M.A.; Korobov, M. Survey study of mercury determination in detonation nanodiamonds by pyrolysis flameless atomic absorption spectroscopy. Diam. Relat. Mater. 2014, 50, 60–65. [Google Scholar] [CrossRef]
- Max, J.J.; Chapados, C. Isotope effects in liquid water by infrared spectroscopy. III. H2O and D2O spectra from 6000 to 0 cm−1. J. Chem. Phys. 2009, 131, 184505. [Google Scholar] [CrossRef] [PubMed]
- Gibson, N.; Shenderova, O.A.; Luo, T.; Moseenkov, S.; Bondar, V.; Puzyr, A.; Purtov, K.; Fitzgerald, Z.; Brenner, D. Colloidal stability of modified nanodiamond particles. Diam. Relat. Mater. 2009, 18, 620–626. [Google Scholar] [CrossRef]
- Chung, K.; Tomljenovic-Hanic, S. Emission Properties of Fluorescent Nanoparticles Determined by Their Optical Environment. Nanomaterials 2015, 5, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Batsanov, S.S.; Dan’Kin, D.A.; Gavrilkin, S.M.; Druzhinina, A.I.; Batsanov, A.S. Structural changes in colloid solutions of nanodiamond. New J. Chem. 2020, 44, 1640–1647. [Google Scholar] [CrossRef]
- Usoltseva, L.O.; Volkov, D.; Nedosekin, D.; Korobov, M.; Proskurnin, M.A.; Zharov, V. Absorption spectra of nanodiamond aqueous dispersions by optical absorption and optoacoustic spectroscopies. Photoacoustics 2018, 12, 55–66. [Google Scholar] [CrossRef]
- Yang, C.Q.; Fateley, W.G. The effect of particle size on fourier-transform infrared photoacoustic spectra. J. Mol. Struct. 1986, 146, 25–39. [Google Scholar] [CrossRef]
- Podobedov, V.B.; Eppeldauer, G.P.; Hanssen, L.M.; Larason, T.C. Calibration of spectral responsivity of IR detectors in the range from 0.6 μm to 24 μm. In Proceedings of the Infrared Technology and Applications XLII, Baltimore, MD, USA, 17–21 April 2016; p. 98190. [Google Scholar] [CrossRef]
- Nguyen, T.; Janik, L.; Raupach, M. Diffuse reflectance infrared fourier transform (DRIFT) spectroscopy in soil studies. Soil Res. 1991, 29, 49–67. [Google Scholar] [CrossRef]
- Deb, P.; Haldar, T.; Kashid, S.M.; Banerjee, S.; Chakrabarty, S.; Bagchi, S. Correlating Nitrile IR Frequencies to Local Electrostatics Quantifies Noncovalent Interactions of Peptides and Proteins. J. Phys. Chem. B 2016, 120, 4034–4046. [Google Scholar] [CrossRef] [Green Version]
- Bégué, D.; Qiao, G.G.; Wentrup, C. Nitrile Imines: Matrix Isolation, IR Spectra, Structures, and Rearrangement to Carbodiimides. J. Am. Chem. Soc. 2012, 134, 5339–5350. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Zhang, Y.; Zhang, Z.; Shan, C.; Shen, W.; Jia, X. Preparation of “natural” diamonds by HPHT annealing of synthetic diamonds. CrystEngComm 2018, 20, 505–511. [Google Scholar] [CrossRef]
- McClelland, J.F.; Jones, R.W.; Bajic, S.J.; Griffiths, P.R. Photoacoustic Spectroscopy. In Handbook of Vibrational Spectroscopy; Chalmers, J.M., Griffiths, P.R., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Calderón, F.J.; Reeves, J.B.; Collins, H.P.; Paul, E.A. Chemical Differences in Soil Organic Matter Fractions Determined by Diffuse-Reflectance Mid-Infrared Spectroscopy. Soil Sci. Soc. Am. J. 2011, 75, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Morterra, C.; Low, M.J.D. The Nature of the 1600 cm−1 Band of Carbons. Spectrosc. Lett. 1982, 15, 689–697. [Google Scholar] [CrossRef]
- Seredych, M.; Rossin, J.A.; Bandosz, T.J. Changes in graphite oxide texture and chemistry upon oxidation and reduction and their effect on adsorption of ammonia. Carbon 2011, 49, 4392–4402. [Google Scholar] [CrossRef]
- Navarro-Pardo, F.; Martínez-Barrera, G.; Martinez-Hernandez, A.L.; Castaño, V.M.; Rivera-Armenta, J.L.; Medellín-Rodríguez, F.J.; Velasco-Santos, C. Effects on the Thermo-Mechanical and Crystallinity Properties of Nylon 6,6 Electrospun Fibres Reinforced with One Dimensional (1D) and Two Dimensional (2D) Carbon. Materials 2013, 6, 3494–3513. [Google Scholar] [CrossRef] [Green Version]
- Bertaux, J.; Froehlich, F.; Ildefonse, P. Multicomponent analysis of FTIR spectra; quantification of amorphous and crystallized mineral phases in synthetic and natural sediments. J. Sediment. Res. 1998, 68, 440–447. [Google Scholar] [CrossRef]
- Brown, D.J.; Shepherd, K.D.; Walsh, M.G.; Mays, M.D.; Reinsch, T.G. Global soil characterization with VNIR diffuse reflectance spectroscopy. Geoderma 2006, 132, 273–290. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product Name | Description | Manufacturer |
|---|---|---|
| RDDM | detonation polycrystalline diamond of RDDM grade, fraction 0–0.125 | “Real-Dzerzhinsk” Ltd., Dzerzhinsk, Russia |
| RUDDM a | nanodiamond material of RUDDM grade, fraction 0–150 | |
| SDND | 5 wt. % Single-Digit NanoDiamonds aqueous suspension | PlasmaChem GmbH, Berlin, Germany |
| PL-D-G | Purified powder grade G | |
| PL-D-G02 | Extra-pure, grade G-02 | |
| PL-D-G01P | agglomerate free, positively charged | |
| PL-Nanopure-G01P | 4 wt.% nanodiamonds aqueous suspension, grade G | |
| DNA-TAN | DNA-TAN | Special Construction-Technological Bureau “Tekhnolog”, St. Petersburg, Russia |
| DNA-STP | DNA-STP | |
| UDA-S | UDA-S, ultradispersed diamond powder | The Laboratory of ultradispersed diamonds of Joint Stock Company Federal Research and Production Center ALTAI, Biysk, Russia |
| UDA-S-GO | UDA-S-GO, ultradipersed diamond powder of deep purification | |
| UDA-SP | UDA-SP, ultradispersed diamonds | JSC “SINTA”, Minsk, Republic of Belarus |
| UDA-GO-SP | UDA-GO-SP, deep purified ultradispersed diamonds | |
| UDA-GO-SP-M1 | UDA-GO-SP-M1, modified ultradispersed diamonds, type M1 | |
| UDA-GO-SP-M2 | UDA-GO-SP-M2, modified ultradispersed diamonds, type M2 |
| Spectral range, cm−1 | 4000–500 |
| Resolution, cm−1 | 4 |
| Background scan | 64; 256 |
| Sample scan | 64; 256 |
| Phase resolution | 10 |
| Phase correction mode | Mertz |
| Zero filling factor | 2 |
| Apodization function | Blackman–Harris 3-Term |
| Aperture setting | 8 mm |
| Interferometer frequency | 1.6; 2.5; 5 kHz |
| Sample and background pre-amplification gain | B (middle amplification) |
| Sample signal gain | Auto |
| Detector | microphone |
| Source | MIR |
| Beam splitter | KBr |
| Spectral range, cm−1 | 4000–370 (with DLaTGS detector) or 6000–700 (with MCT detector) |
| Resolution, cm−1 | 2 |
| Background scan | 128 |
| Sample scan | 128 |
| Aperture setting | 8 mm |
| Phase resolution | 4 |
| Phase correction mode | Mertz |
| Zero filling factor | 1 |
| Apodization function | Blackman–Harris 3-Term |
| Sample and background pre-amplification gain | “Ref” (without amplification) |
| Background signal gain | Auto |
| Sample signal gain | Auto |
| Scanner velocity | 10 kHz |
| Detector | Room temperature DLaTGS or liquid nitrogen cooled photovoltaic MCT |
| Source | MIR |
| Beam splitter | KBr |
| Background | Diamond crystal with a lowered pressure screw with a flat end |
| Spectral range, cm−1 | 7000–400 |
| Resolution, cm−1 | 2 |
| Background scan | 256 |
| Sample scan | 256 |
| Phase resolution | 16 |
| Phase correction mode | Mertz |
| Zero filling factor | 2 |
| Apodization function | Blackman–Harris 3-Term |
| Aperture setting | 3 mm |
| Sample and background pre-amplification gain | “Ref” (without amplification) for DLaTGS detector A (standard amplification) for MCT detector |
| Background signal gain | Auto |
| Sample signal gain | Auto |
| Scanner velocity | 10 kHz |
| Detector | Room temperature DLaTGS or liquid nitrogen cooled photovoltaic MCT |
| Source | MIR |
| Beam splitter | KBr |
| Background | Mirror |
| Wavenumber | Assignment | ATR | DRIFT | PAS * |
|---|---|---|---|---|
| 5900–5600 | 2ν C–H aliphatic chain stretching | — | W | — |
| 5300 | Water combination band av1 + v2 + bv3; a + b = 1 | — | Mb | Wb |
| 4800 | Aromatic C–H combination bands (?) ** | — | Wb | — |
| 4500–4100 | Aliphatic C–H combination bands | — | Wb To Mb | W |
| 3715 | Hydrogen-bonded –O–H···H–O– stretch | — | Wp | Wp |
| 3695 | Hydrogen-bonded –O–H···H2O stretch | — | Mp | Wp (noisy) |
| 3569 | Hydrogen-bonded RO–H···H2O H–OR stretch (?) | — | W | W (noisy) |
| 3450–3420 | Liquid: antisynchronous stretch v3 | Mv | Sv | Mv |
| 3407 | O–H stretch and intermolecular hydrogen bonds (unresolved) | Mb to Wb | — | Mb to Wb |
| 3290 | H–O–H bend of liquid adsorbed water, 2v2 | — | Wv | W |
| 3230–3210 | Liquid: synchronous stretch, v1 | Sv | Sv | Sv |
| 3050 | Aromatic C–H stretching | W to none | S | M |
| 2970 | Alkene C–H stretch | W to none | M | W |
| 2950–2940 | Aliphatic C–H, CH3 antisymmetric stretch | W to none | S | M |
| 2940–2930 | Aliphatic C–H, CH2 antisymmetric stretch | — | S | S |
| 2880–2870 | Aliphatic C–H, CH3 symmetric stretch | W to none | S | M |
| 2850–2835 | Aliphatic C–H, CH2 symmetric stretch | W to none | Wb | M |
| 2750–2550 | Carboxylic O–H stretch | Wb to none | Mb to Wb | Wb to none* |
| 2150 | Water combination band v2 + L2 | Wb | Wb | Wb |
| 1800–1780 | C=O stretch of conjugated carboxyl groups | M | S | S to M * |
| 1765–1730 | C=O stretch of monomeric carboxyl groups | Mb | Sb | Sb to Mb * |
| 1670 | C=O stretch of non-carboxyl carbonyl (?) C=C stretch | W | W | M |
| 1644–1642 | H–O–H bend of liquid water, v2 | Sv | Mv | Mv |
| 1630–1625 | H–O–H bend of adsorbed liquid water, v2 | Sv | Mv | Mv |
| 1610 | H–O–H bend of adsorbed liquid water, v2 | Ssh | Msh | Msh |
| 1580–1560 | adsorbed water, C=C stretch, (?) | M | M | M |
| 1470–1450 | sp3 CH2 wagging | S | M | |
| 1440 | Carboxyl C–O–H in-plane bend Aromatic, ring C=C stretch | — | Wb | W |
| 1410 | Carboxyl C–O–H in-plane bend | Msh | Msh | Msh |
| 1400–1395 | Non-carboxyl C–O–H in-plane bend CH2 deformation (scissors) | Msh | Ssh | Ssh |
| 1373 | Non-carboxyl C–O–H in-plane bend CH3 deformation (umbrella) | M | Ssh | M |
| 1330 | Non-carboxyl C–O–H in-plane bend (?) | S | M | S to M * |
| 1270–1267 | Carboxyl C–O stretch | W | M | M |
| 1245–1235 | C–N stretch | W | W | W |
| 1192 | C–C–C (?) | — | M | W |
| 1145–1130 | C–O–C (?) | — | M | W |
| 1103 | Non-carboxyl C–O stretch | Sb | Sb | Sb |
| 1060–1040 | In plane –C–H bend (non-aromatic) and carbohydrates (?) | W | Wsh | Wb |
| 1000–500 | Water librations, L2 | Sb | Sb | Mb to Wb |
| 960–940 | Carboxyl out-of-plane C–O–H bend, =CH2 wagging (?) | M | Wb | M to W * |
| 830 | Aromatic =C–H bend | Mp | Mp | Mp |
| 760 | Polyaromatic =C–H bend (?) | — | W | M to none * |
| 610 | Non-carboxyl out-of-plane C–O–H bend | W | M | Wb to none * |
| 410 | (?) C–C in-phase vibrations | M | Wb | — |
| Band Center, cm−1 | High-Wave Boundary, cm−1 | Low-Wave Boundary, cm−1 | RSD |
|---|---|---|---|
| 2935 | 2952 | 2918 | 0.27 |
| 2837 | 2857 | 2817 | 0.25 |
| 2650 | 2673 | 2626 | 0.43 |
| 1750 | 1815 | 1684 | 0.21 |
| 1630 | 1668 | 1535 | 0.18 |
| 1400 | 1417 | 1393 | 0.20 |
| 1270 | 1341 | 1249 | 0.21 |
| Band Center, cm−1 | High-Wave Boundary, cm−1 | Low-Wave Boundary, cm−1 | RSD |
|---|---|---|---|
| 2935 | 2952 | 2918 | 0.40 |
| 2837 | 2857 | 2817 | 0.40 |
| 2650 | 2677 | 2622 | 0.27 |
| 1750 | 1852 | 1691 | 0.33 |
| 1630 | 1690 | 1594 | 0.32 |
| 1400 | 1417 | 1393 | 0.33 |
| 1270 | 1341 | 1244 | 0.39 |
| Band Center, cm−1 | High-Wave Boundary, cm−1 | Low-Wave Boundary, cm−1 | RSD |
|---|---|---|---|
| 2935 | 2952 | 2918 | 0.34 |
| 2837 | 2857 | 2817 | 0.37 |
| 2650 | 2676 | 2619 | 0.23 |
| 1750 | 1852 | 1691 | 0.10 |
| 1630 | 1690 | 1594 | 0.08 |
| 1400 | 1417 | 1393 | 0.11 |
| 1270 | 1341 | 1253 | 0.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volkov, D.S.; Krivoshein, P.K.; Proskurnin, M.A. Detonation Nanodiamonds: A Comparison Study by Photoacoustic, Diffuse Reflectance, and Attenuated Total Reflection FTIR Spectroscopies. Nanomaterials 2020, 10, 2501. https://doi.org/10.3390/nano10122501
Volkov DS, Krivoshein PK, Proskurnin MA. Detonation Nanodiamonds: A Comparison Study by Photoacoustic, Diffuse Reflectance, and Attenuated Total Reflection FTIR Spectroscopies. Nanomaterials. 2020; 10(12):2501. https://doi.org/10.3390/nano10122501
Chicago/Turabian StyleVolkov, Dmitry S., Petr K. Krivoshein, and Mikhail A. Proskurnin. 2020. "Detonation Nanodiamonds: A Comparison Study by Photoacoustic, Diffuse Reflectance, and Attenuated Total Reflection FTIR Spectroscopies" Nanomaterials 10, no. 12: 2501. https://doi.org/10.3390/nano10122501