
PEGylated Paclitaxel Nanomedicine Meets 3D Confinement: Cytotoxicity and Cell Behaviors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
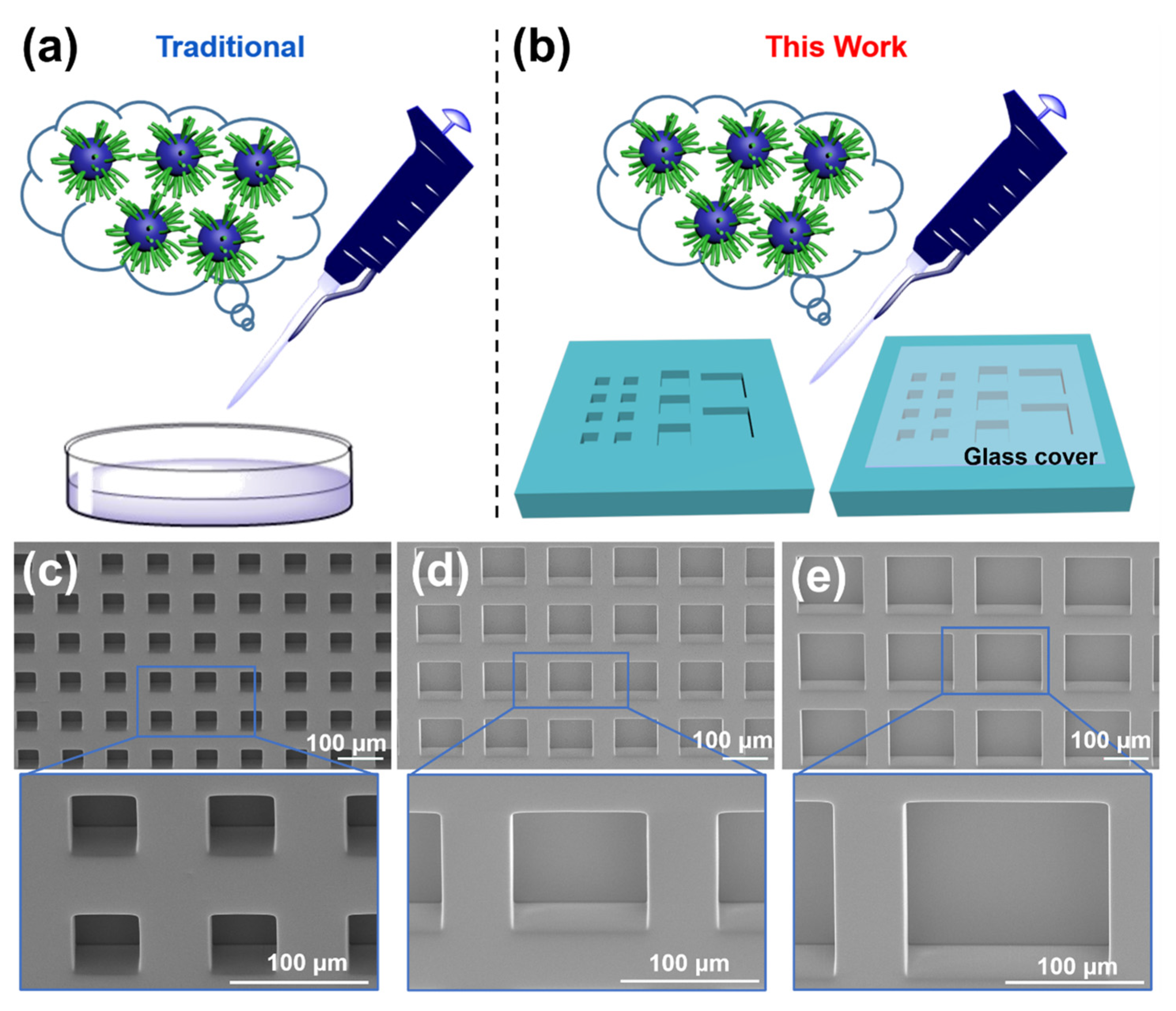
2.3. Fabrication of Microwell Arrays
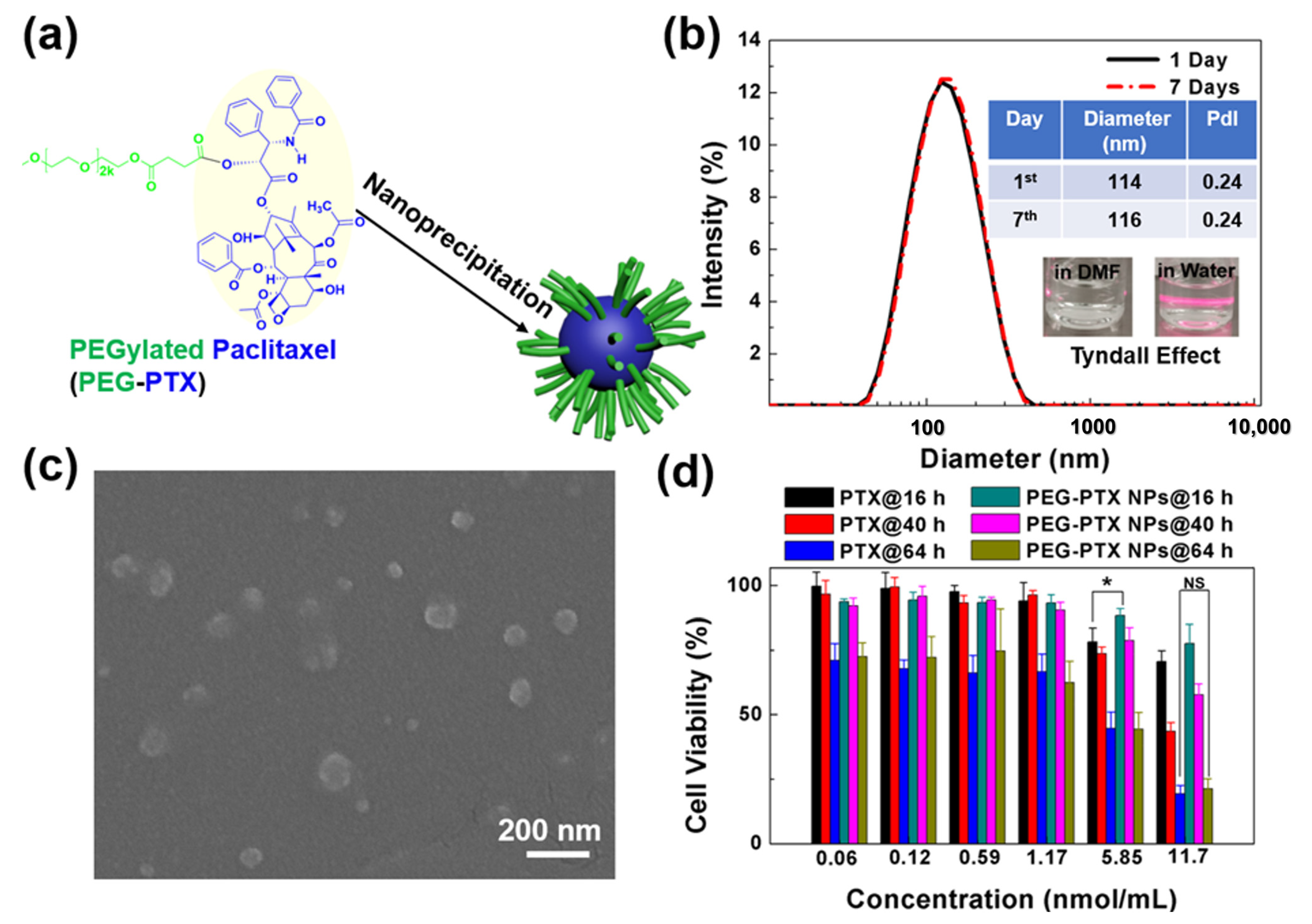
2.4. Synthesis of PEG-PTX NPs
2.5. Cell Culture
2.6. Cell Viability Assays
2.7. Time-Lapse Imaging
2.8. Cell Preparation for Scanning Electron Microscopy
3. Results and Discussions
3.1. Characterization of Microwells and PEG-PTX NPs
3.2. Cytotoxicity of PEG-PTX NPs in Microwells
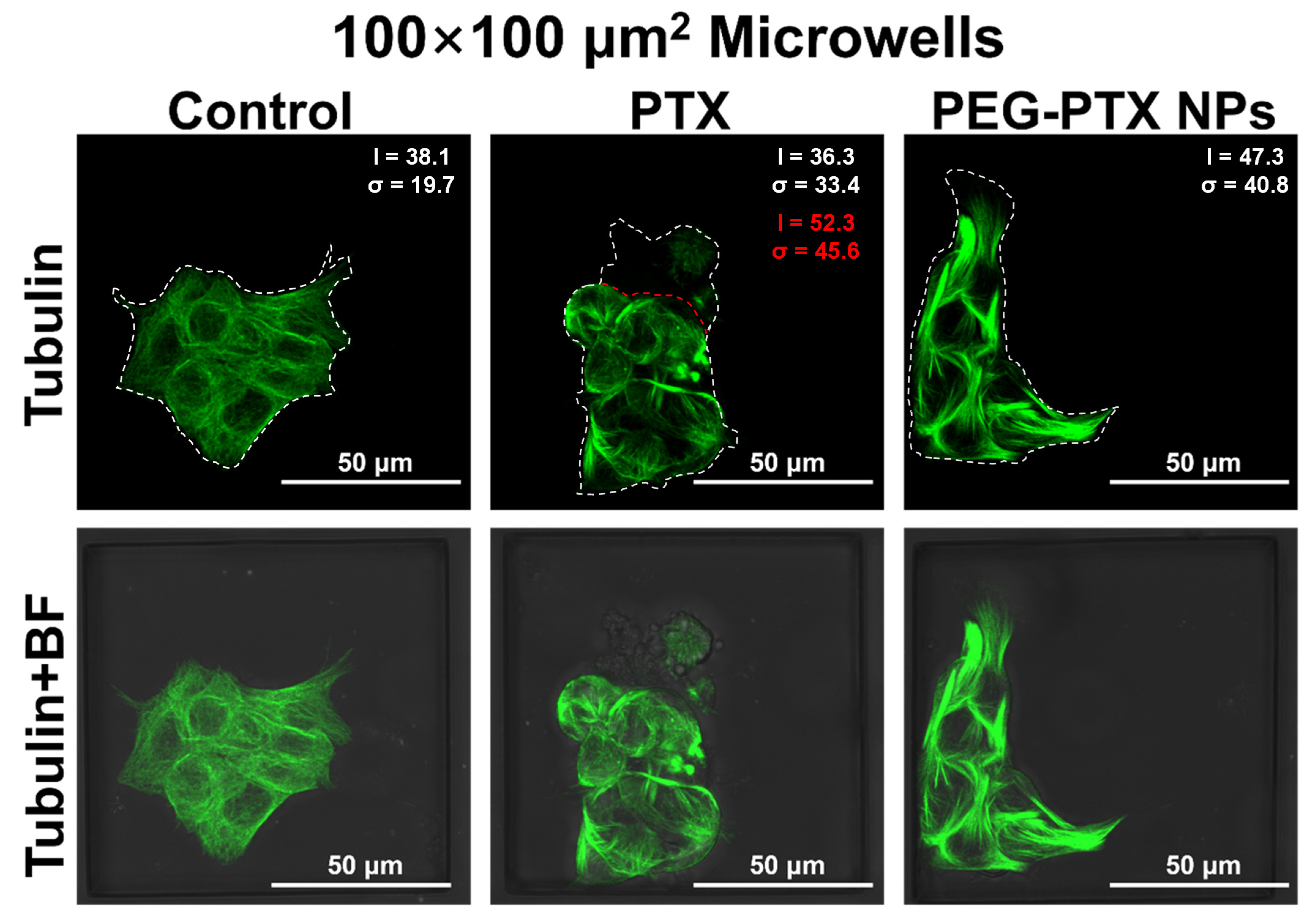
3.2.1. PEG-PTX NPs Followed Same Working Mechanism in Microwells as on Open Regions
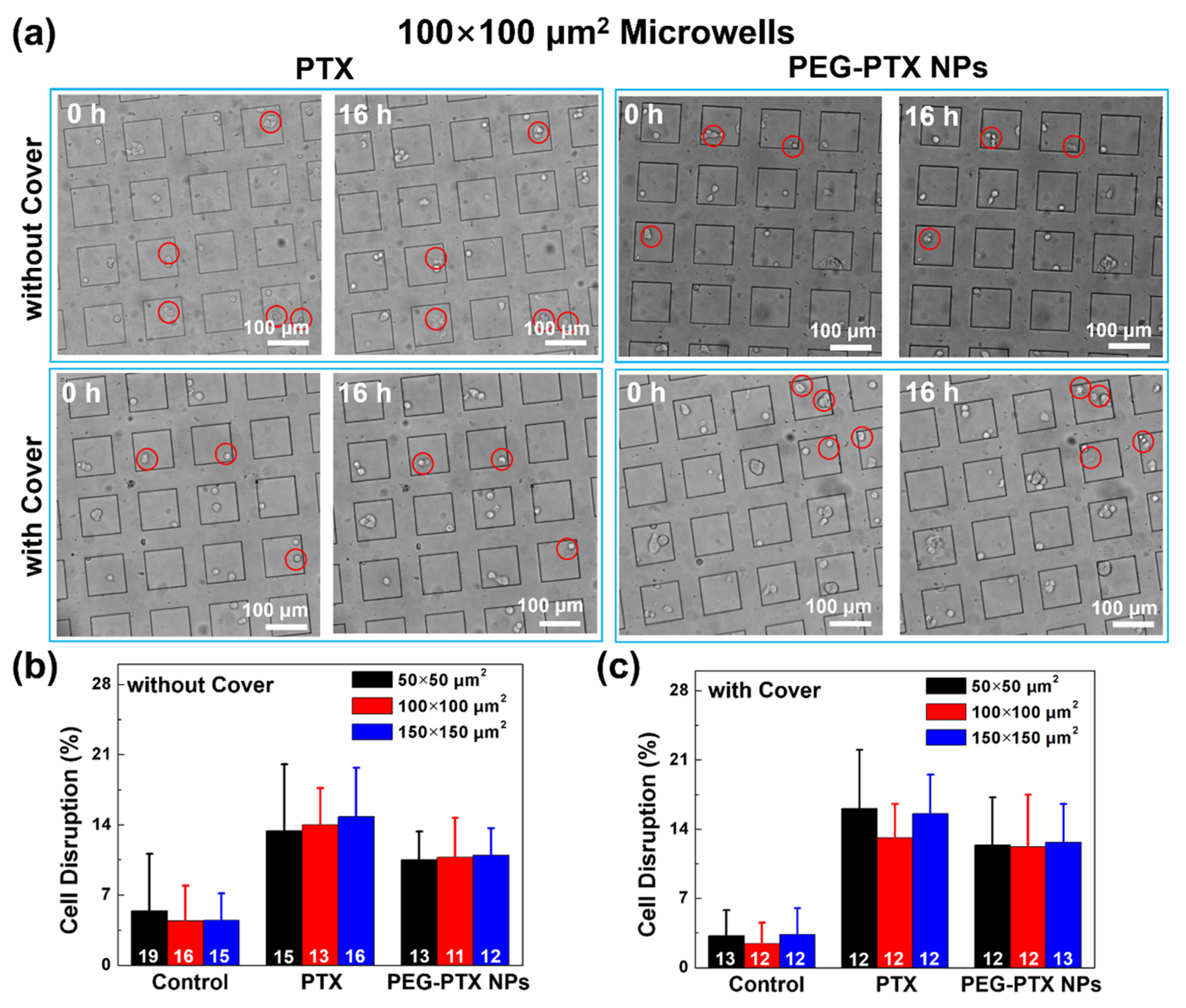
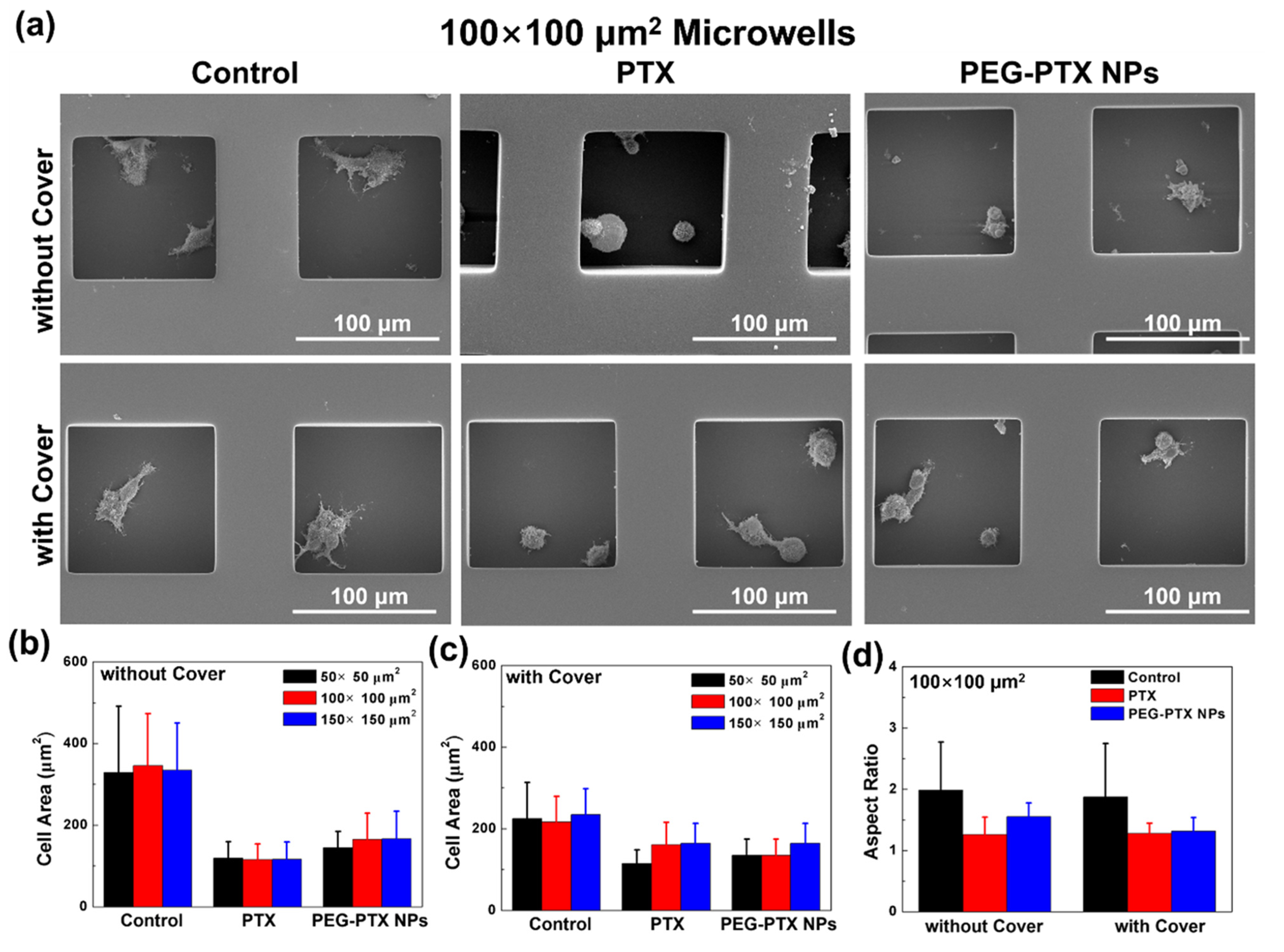
3.2.2. Reduced Overall Cytotoxicity with Microwell Confinement and Isolation
3.3. NPC43 Cell Behaviors with PTX and PEG-PTX NPs Treatments
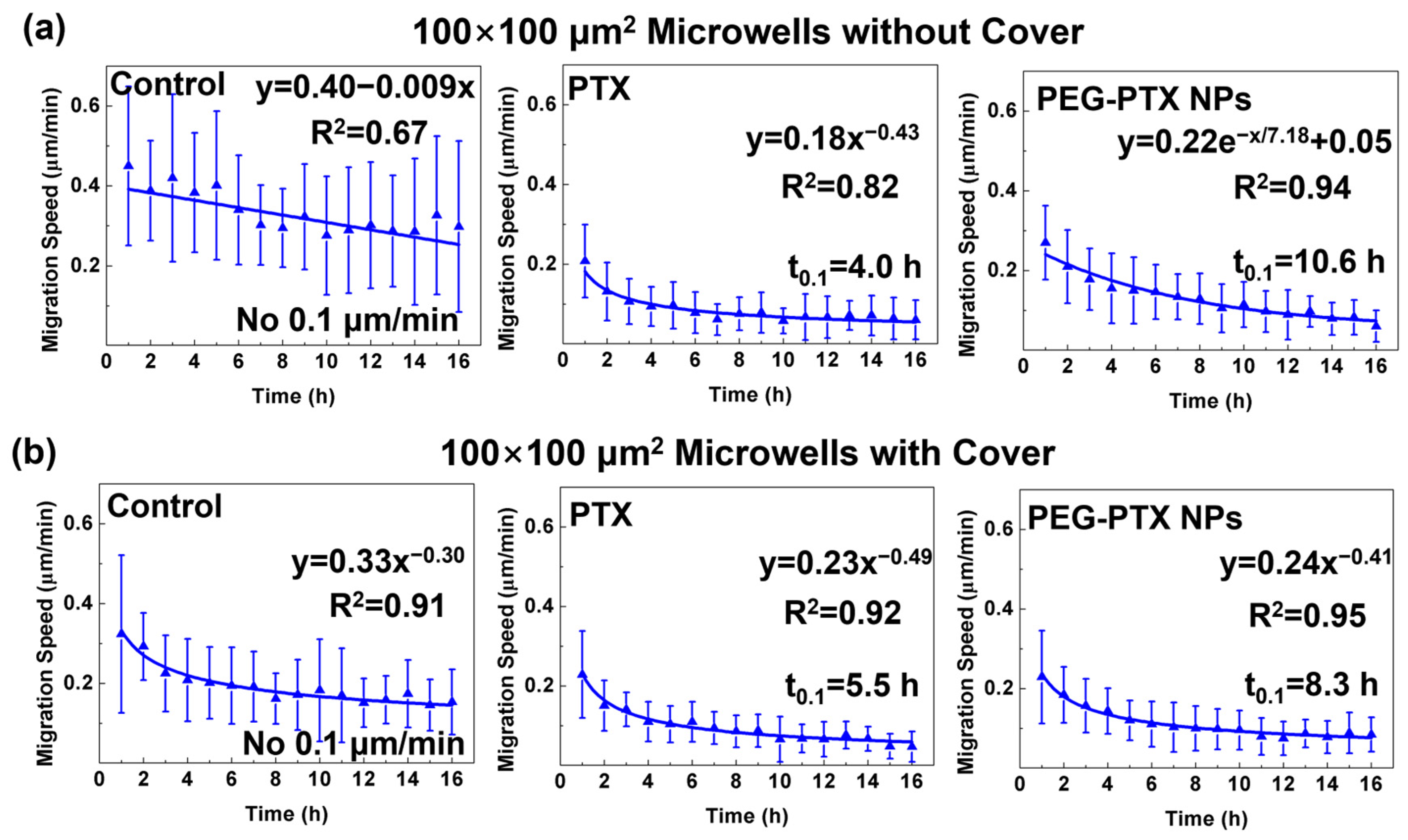
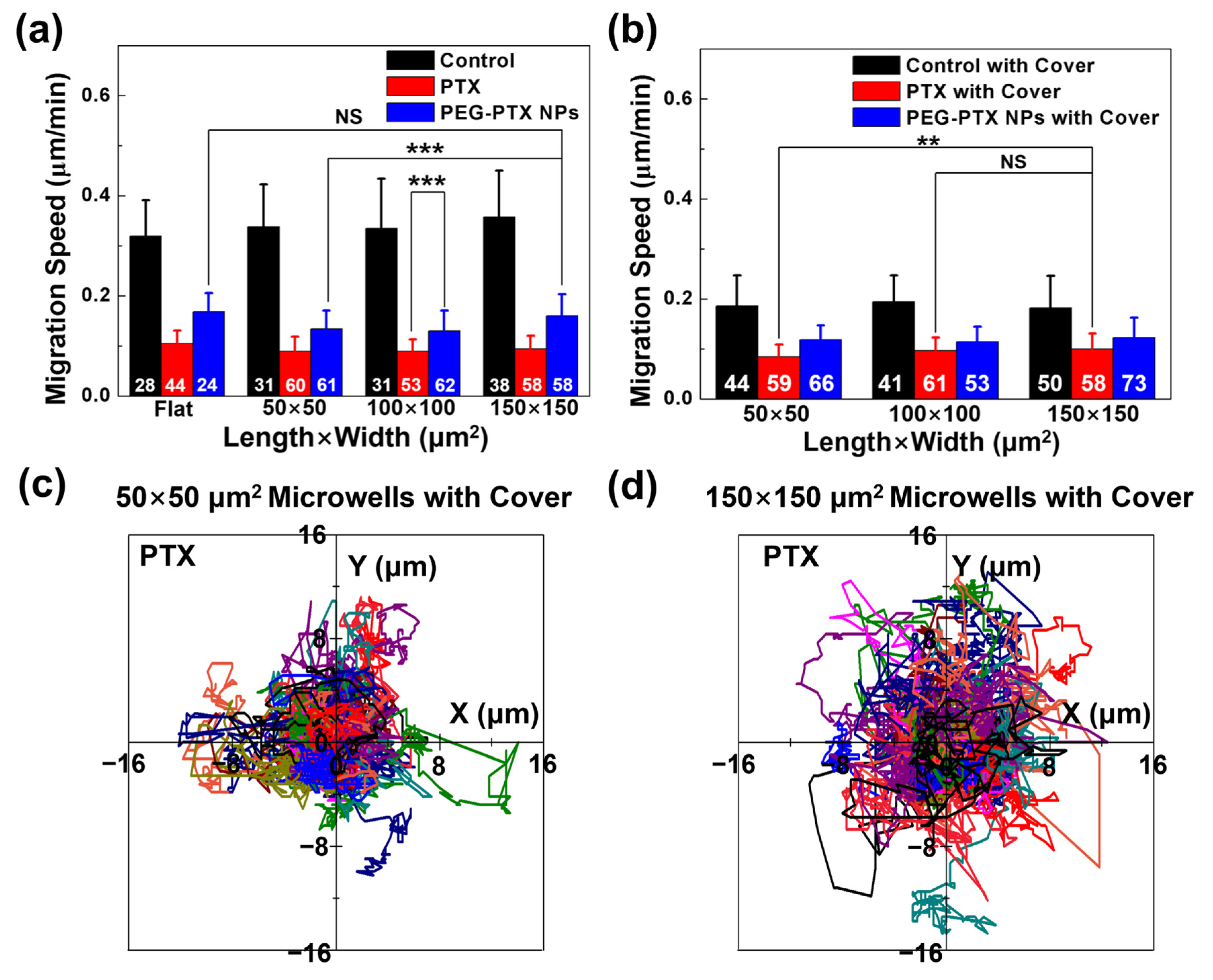
3.3.1. Reduced NPC43 Migration Speed over Time with Drug Treatment
3.3.2. PTX Took Effect Faster than PEG-PTX NPs under Microwell Confinement
3.3.3. PEG-PTX NPs Had More Stable Performance than PTX under Isolation and Confinement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kargozar, S.; Mollazadeh, S.; Kermani, F.; Webster, T.J.; Nazarnezhad, S.; Hamzehlou, S.; Baino, F. Hydroxyapatite nanoparticles for improved cancer theranostics. J. Funct. Biomater. 2022, 13, 100. [Google Scholar] [CrossRef] [PubMed]
- Crawley, J.W.; Gow, I.E.; Lawes, N.; Kowalec, I.; Kabalan, L.; Catlow, C.R.A.; Logsdail, A.J.; Taylor, S.H.; Dummer, N.F.; Hutchings, G.J. Heterogeneous trimetallic nanoparticles as catalysts. Chem. Rev. 2022, 122, 6795–6849. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Colombani-Garay, D.; Huang, L.; Duan, C.; Han, G. Tailoring nanoparticles based on boron dipyrromethene for cancer imaging and therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1627. [Google Scholar] [CrossRef]
- Song, D.; Xu, Q. Engineering a Nano/Biointerface for Cell and Organ-Selective Drug Delivery. Langmuir 2022, 38, 9092–9098. [Google Scholar] [CrossRef] [PubMed]
- Calzoni, E.; Cesaretti, A.; Polchi, A.; Di Michele, A.; Tancini, B.; Emiliani, C. Biocompatible polymer nanoparticles for drug delivery applications in cancer and neurodegenerative disorder therapies. J. Funct. Biomater. 2019, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- De Lázaro, I.; Mooney, D.J. Obstacles and opportunities in a forward vision for cancer nanomedicine. Nat. Mater. 2021, 20, 1469–1479. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Nie, G. Multifunctional biomolecule nanostructures for cancer therapy. Nat. Rev. Mater. 2021, 6, 766–783. [Google Scholar] [CrossRef]
- Lee, E.S.; Shin, J.M.; Son, S.; Ko, H.; Um, W.; Song, S.H.; Lee, J.A.; Park, J.H. Recent advances in polymeric nanomedicines for cancer immunotherapy. Adv. Healthc. Mater. 2019, 8, 1801320. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Meng, Q.; Li, B.; Liu, Y.; Li, Z.; Li, X.; Sun, Z.; Chen, Y. Supramolecular Combination Chemotherapy: Cucurbit[8]uril Complex Enhanced Platinum Drug Infiltration and Modified Nanomechanical Property of Colorectal Cancer Cells. Langmuir 2022, 38, 14326–14334. [Google Scholar] [CrossRef]
- Min, Y.; Caster, J.M.; Eblan, M.J.; Wang, A.Z. Clinical translation of nanomedicine. Chem. Rev. 2015, 115, 11147–11190. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, N.K.; Desai, N.; Legha, S.; Soon-Shiong, P.; Theriault, R.L.; Rivera, E.; Esmaeli, B.; Ring, S.E.; Bedikian, A.; Hortobagyi, G.N. Phase I and pharmacokinetic study of ABI-007, a Cremophor-free, protein-stabilized, nanoparticle formulation of paclitaxel. Clin. Cancer Res. 2002, 8, 1038–1044. [Google Scholar]
- Desai, N.; Trieu, V.; Yao, Z.; Louie, L.; Ci, S.; Yang, A.; Tao, C.; De, T.; Beals, B.; Dykes, D. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin. Cancer Res. 2006, 12, 1317–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.-Y.; Kim, D.-W.; Chung, J.-Y.; Shin, S.G.; Kim, S.-C.; Heo, D.S.; Kim, N.K.; Bang, Y.-J. Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin. Cancer Res. 2004, 10, 3708–3716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.C.; Kim, D.W.; Shim, Y.H.; Bang, J.S.; Oh, H.S.; Kim, S.W.; Seo, M.H. In vivo evaluation of polymeric micellar paclitaxel formulation: Toxicity and efficacy. J. Control. Release 2001, 72, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-E.; Kim, H.; Doh, J. Single cell arrays of hematological cancer cells for assessment of lymphocyte cytotoxicity dynamics, serial killing, and extracellular molecules. Lab Chip 2019, 19, 2009–2018. [Google Scholar] [CrossRef]
- Xia, Y.; Chen, H.; Li, J.; Hu, H.; Qian, Q.; He, R.-X.; Ding, Z.; Guo, S.-S. Acoustic Droplet-Assisted Superhydrophilic–Superhydrophobic Microarray Platform for High-Throughput Screening of Patient-Derived Tumor Spheroids. ACS Appl. Mater. Interfaces 2021, 13, 23489–23501. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Guo, J.; Chen, H.; Zhang, D.; Shang, L.; Zhang, B.; Zhao, Y. Tailoring materials with specific wettability in biomedical engineering. Adv. Sci. 2021, 8, 2100126. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Chen, L.; Wei, W.; Qian, K.; Liu, Y.; Leng, J. Application and development of shape memory micro/nano patterns. Small 2022, 18, 2105958. [Google Scholar] [CrossRef]
- Chen, Z.; Kheiri, S.; Young, E.W.; Kumacheva, E. Trends in droplet microfluidics: From droplet generation to biomedical applications. Langmuir 2022, 38, 6233–6248. [Google Scholar] [CrossRef]
- Tu, T.Y.; Wang, Z.; Bai, J.; Sun, W.; Peng, W.K.; Huang, R.Y.J.; Thiery, J.P.; Kamm, R.D. Rapid prototyping of concave microwells for the formation of 3D multicellular cancer aggregates for drug screening. Adv. Healthc. Mater. 2014, 3, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Sui, X.; Song, F.; Li, Y.; Li, K.; Chen, Z.; Yang, F.; Chen, X.; Zhang, Y.; Wang, X. Lung cancer organoids analyzed on microwell arrays predict drug responses of patients within a week. Nat. Commun. 2021, 12, 2581. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Suhito, I.R.; Angeline, N.; Han, Y.; Son, H.; Luo, Z.; Kim, T.H. Vertically Coated Graphene Oxide Micro-Well Arrays for Highly Efficient Cancer Spheroid Formation and Drug Screening. Adv. Healthc. Mater. 2020, 9, 1901751. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Zhang, J.; Wu, M.; Zhang, Y. A dynamic 3D tumor spheroid chip enables more accurate nanomedicine uptake evaluation. Adv. Sci. 2019, 6, 1901462. [Google Scholar] [CrossRef] [PubMed]
- Kang, A.; Seo, H.I.; Chung, B.G.; Lee, S.-H. Concave microwell array-mediated three-dimensional tumor model for screening anticancer drug-loaded nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1153–1161. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, S.; Lam, Y.W.; Pang, S.W. Dynamics of natural killer cells cytotoxicity in microwell arrays with connecting channels. Front. Immunol. 2017, 8, 998. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, Y.J.; Berger, C.T.; Sips, M.; Cheney, P.C.; Alter, G.; Love, J.C. Single-cell analysis of the dynamics and functional outcomes of interactions between human natural killer cells and target cells. Integr. Biol. 2012, 4, 1175–1184. [Google Scholar] [CrossRef] [Green Version]
- Gui, G.; Fan, Z.; Ning, Y.; Yuan, C.; Zhang, B.; Xu, Q. Optimization, characterization and in vivo evaluation of paclitaxel-loaded folate-conjugated superparamagnetic iron oxide nanoparticles. Int. J. Nanomed. 2021, 16, 2283. [Google Scholar] [CrossRef]
- Li, X.; Lu, W.; Zhou, T.; Zhao, F.; Yang, L. Timosaponin AIII Suppresses RAP1 Signaling Pathway to Enhance the Inhibitory Effect of Paclitaxel on Nasopharyngeal Carcinoma. Comput. Math. Methods Med. 2022, 2022, 6756676. [Google Scholar] [CrossRef]
- Bian, S.; Wang, Z.; Chen, Y.; Li, R. SPLUNC1 and MLL3 regulate cancer stem cells in nasopharyngeal carcinoma. J. Buon 2019, 24, 1700–1705. [Google Scholar]
- Aaro, T.; Jaana, R.; Reidar, G.; Kari, S.; Stina, S. Epstein-Barr virus (EBV)-encoded small RNAs (EBERs) associated with poor prognosis of head and neck carcinomas. Oncotarget 2017, 8, 27328. [Google Scholar]
- Xu, Y.; Pang, S.W. Natural killer cell migration control in microchannels by perturbations and topography. Lab Chip 2019, 19, 2466–2475. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Sun, T.; Xie, Z.; Gu, J.; Jing, X. A dual-responsive nanocapsule via disulfide-induced self-assembly for therapeutic agent delivery. Chem. Sci. 2016, 7, 1846–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Lin, W.; Zhang, W.; Feng, Y.; Wu, Z.; Chen, L.; Xie, Z. PEGylated BODIPY assembling fluorescent nanoparticles for photodynamic therapy. Chin. Chem. Lett. 2017, 28, 1875–1877. [Google Scholar] [CrossRef]
- Lin, W.; Zhang, W.; Liu, S.; Li, Z.; Hu, X.; Xie, Z.; Duan, C.; Han, G. Engineering pH-responsive BODIPY nanoparticles for tumor selective multimodal imaging and phototherapy. ACS Appl. Mater. Interfaces 2019, 11, 43928–43935. [Google Scholar] [CrossRef]
- Lin, W.; Yin, L.; Sun, T.; Wang, T.; Xie, Z.; Gu, J.; Jing, X. The effect of molecular structure on cytotoxicity and antitumor activity of PEGylated nanomedicines. Biomacromolecules 2018, 19, 1625–1634. [Google Scholar] [CrossRef]
- Lin, W.; Guan, X.; Sun, T.; Huang, Y.; Jing, X.; Xie, Z. Reduction-sensitive amphiphilic copolymers made via multi-component Passerini reaction for drug delivery. Colloids Surf. B 2015, 126, 217–223. [Google Scholar] [CrossRef]
- Lu, J.; Chuan, X.; Zhang, H.; Dai, W.; Wang, X.; Wang, X.; Zhang, Q. Free paclitaxel loaded PEGylated-paclitaxel nanoparticles: Preparation and comparison with other paclitaxel systems in vitro and in vivo. Int. J. Pharm. 2014, 471, 525–535. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Q.; Zhang, Z.; Tong, J.; Chu, D.; Gu, J. Simultaneous quantitative analysis of polyethylene glycol (PEG), PEGylated paclitaxel and paclitaxel in rats by MS/MSALL technique with hybrid quadrupole time-of-flight mass spectrometry. J. Pharm. Biomed. Anal. 2017, 145, 255–261. [Google Scholar] [CrossRef]
- Zhang, H.; Hu, H.; Zhang, H.; Dai, W.; Wang, X.; Wang, X.; Zhang, Q. Effects of PEGylated paclitaxel nanocrystals on breast cancer and its lung metastasis. Nanoscale 2015, 7, 10790–10800. [Google Scholar] [CrossRef]
- Leong, D.T.; Ng, K.W. Probing the relevance of 3D cancer models in nanomedicine research. Adv. Drug Deliv. Rev. 2014, 79, 95–106. [Google Scholar] [CrossRef]
- Boix-Montesinos, P.; Soriano-Teruel, P.M.; Arminan, A.; Orzáez, M.; Vicent, M.J. The past, present, and future of breast cancer models for nanomedicine development. Adv. Drug Deliv. Rev. 2021, 173, 306–330. [Google Scholar] [CrossRef]
- Tang, S.; Yin, Q.; Su, J.; Sun, H.; Meng, Q.; Chen, Y.; Chen, L.; Huang, Y.; Gu, W.; Xu, M. Inhibition of metastasis and growth of breast cancer by pH-sensitive poly (β-amino ester) nanoparticles co-delivering two siRNA and paclitaxel. Biomaterials 2015, 48, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, Z.; Yang, S.; Wang, Y.; Luan, Z. CircEXOC6B suppresses the proliferation and motility and sensitizes ovarian cancer cells to paclitaxel through miR-376c-3p/FOXO3 axis. Cancer Biother. Radiopharm. 2022, 37, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Xu, Y.; Pang, S.W. Enhanced motility and interaction of nasopharyngeal carcinoma with epithelial cells in confined microwells. Lab Chip 2023, 23, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Hutmacher, D.W. Biomaterials offer cancer research the third dimension. Nat. Mater. 2010, 9, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Mandal, M.; Hutmacher, D.W.; Russell, P.J.; Soekmadji, C.; Kundu, S.C. Engineered silk fibroin protein 3D matrices for in vitro tumor model. Biomaterials 2011, 32, 2149–2159. [Google Scholar] [CrossRef]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T. Comparison of 2D-and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Lilly, G.D.; Doty, R.C.; Podsiadlo, P.; Kotov, N.A. In vitro toxicity testing of nanoparticles in 3D cell culture. Small 2009, 5, 1213–1221. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yang, X.; Zou, J.; Jia, C.; Hu, Y.; Du, H.; Wang, H. Evaluation of photodynamic therapy efficiency using an in vitro three-dimensional microfluidic breast cancer tissue model. Lab Chip 2015, 15, 735–744. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, W.; Xu, Y.; Hong, X.; Pang, S.W. PEGylated Paclitaxel Nanomedicine Meets 3D Confinement: Cytotoxicity and Cell Behaviors. J. Funct. Biomater. 2023, 14, 322. https://doi.org/10.3390/jfb14060322
Lin W, Xu Y, Hong X, Pang SW. PEGylated Paclitaxel Nanomedicine Meets 3D Confinement: Cytotoxicity and Cell Behaviors. Journal of Functional Biomaterials. 2023; 14(6):322. https://doi.org/10.3390/jfb14060322
Chicago/Turabian StyleLin, Wenhai, Yuanhao Xu, Xiao Hong, and Stella W. Pang. 2023. "PEGylated Paclitaxel Nanomedicine Meets 3D Confinement: Cytotoxicity and Cell Behaviors" Journal of Functional Biomaterials 14, no. 6: 322. https://doi.org/10.3390/jfb14060322