

Changes in the Bacterial Community Composition of Cultivated Soil after Digging up Operations for Laying a Pipeline
 ,
,  , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Site Description and Sampling
2.2. The 16S rDNA Amplicon Sequencing and Sequence Processing
2.3. Bioinformatics Analysis
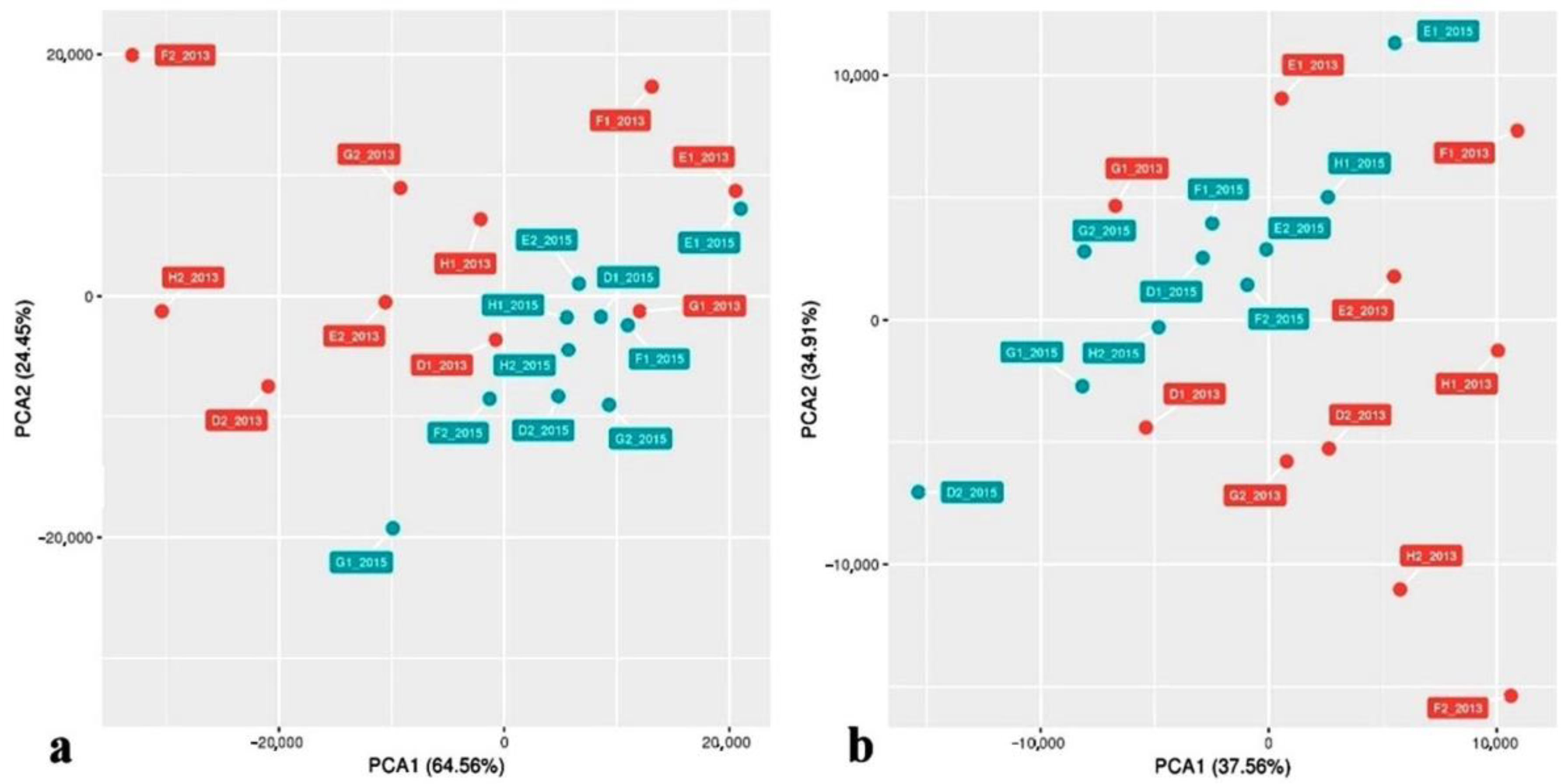
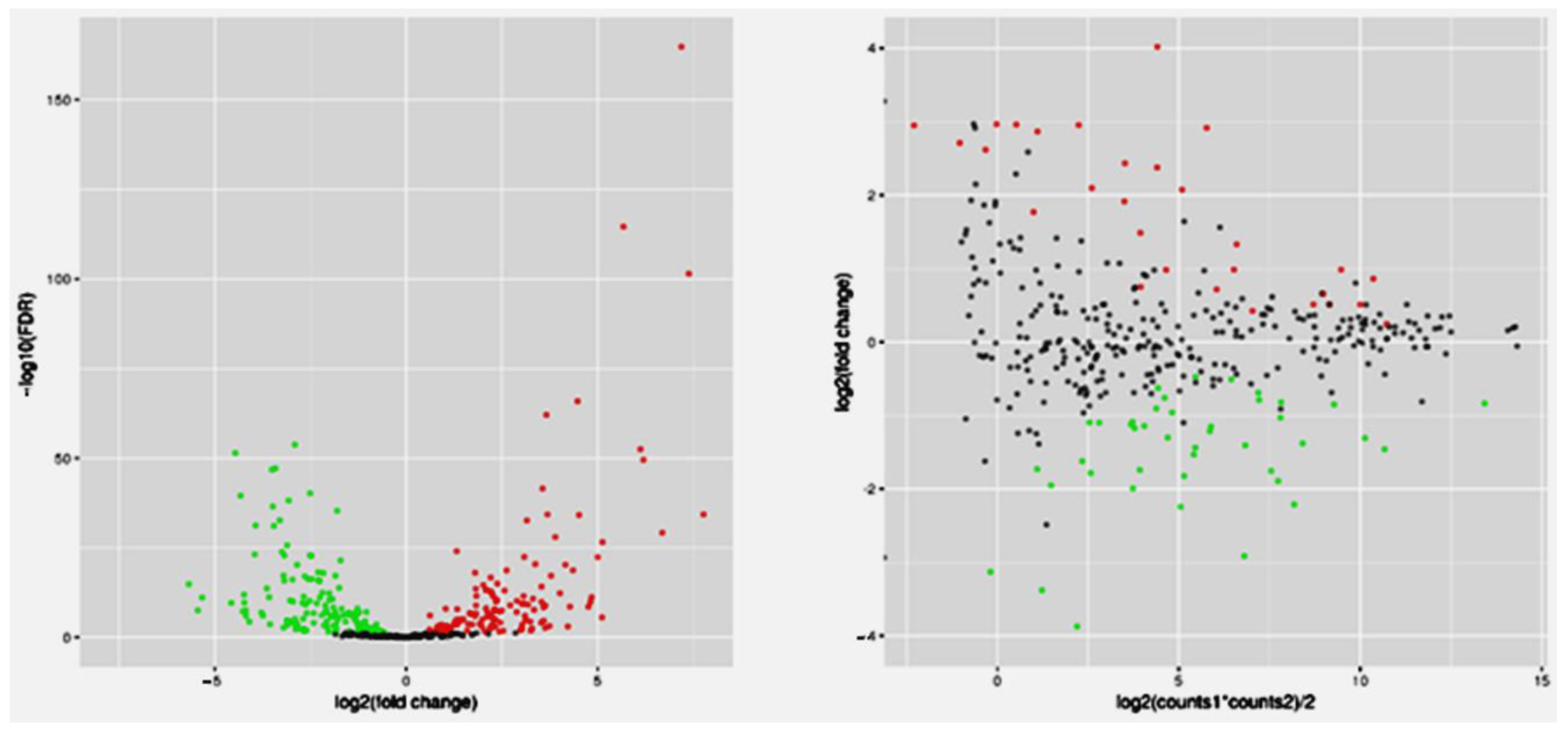
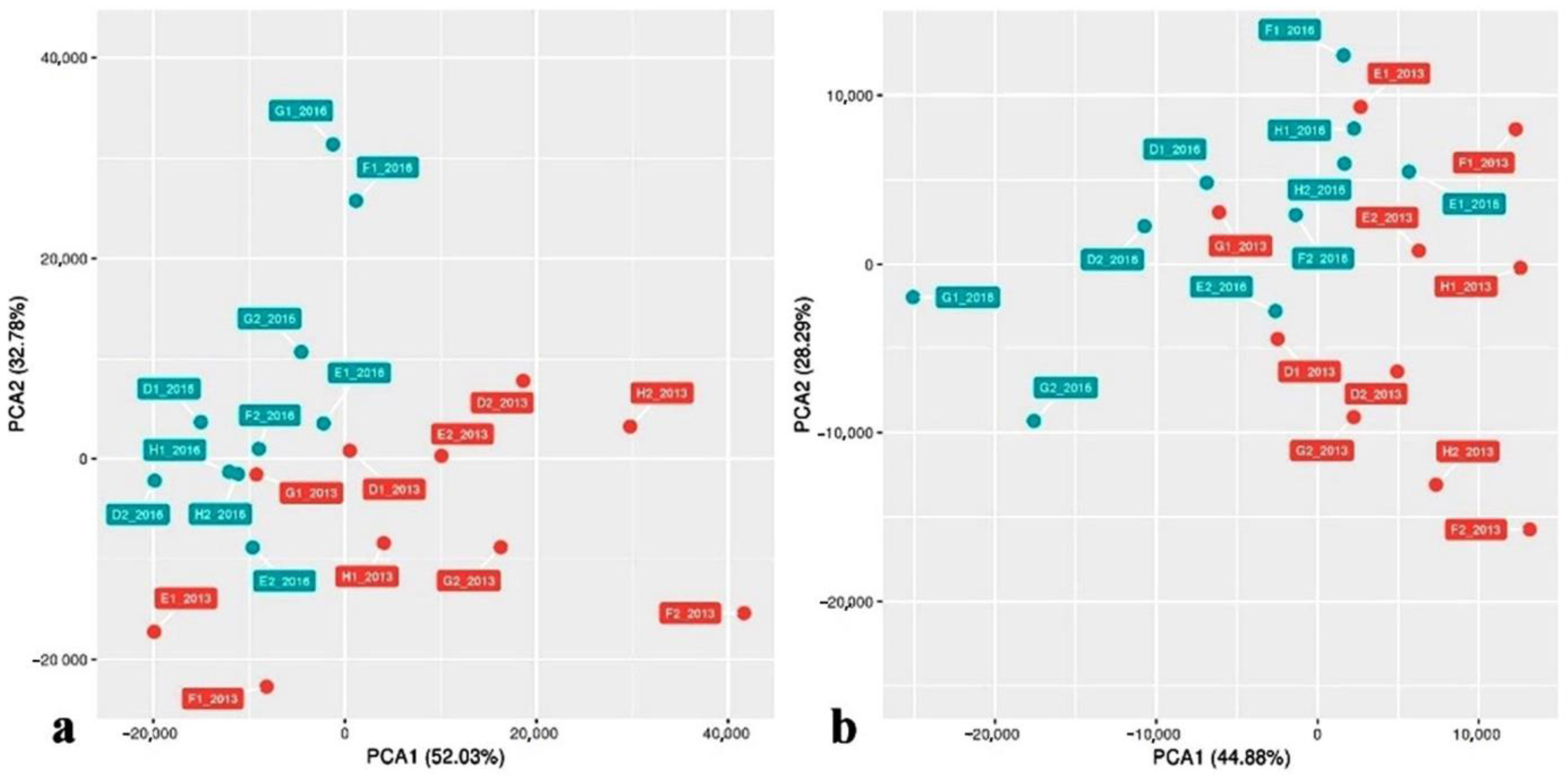
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mocali, S.; Benedetti, A. Exploring research frontiers in microbiology: The challenge of metagenomics in soil microbiology. Res. Microbiol. 2010, 161, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, V.; Verma, M.K.; Gupta, S.; Mandhan, V.; Chauhan, N.S. Metagenomic profiling of soil microbes to mine salt stress tolerance genes. Front. Microbiol. 2018, 9, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azarbad, H.; Niklinska, M.; Laskowski, R.; van Straalen, N.M.; van Gestel, C.A.M.; Zhou, J. Microbial community composition and functions are resilient to metal pollution along two forest soil. FEMS Microbiol. Ecol. 2015, 91, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Meng, D.; Li, J.; Yin, H.; Liu, H.; Liu, X. Response of soil microbial communities and microbial interactions to long-term heavy metal contamination. Environ. Pollut. 2017, 231, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Beattie, R.E.; Henke, W.; Campa, M.F.; Hazen, T.C.; McAliley, L.R.; Campbell, J.H. Variation in microbial community structure correlates with heavy-metal contamination in soils decades after mining ceased. Soil. Biol. Biochem. 2018, 126, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Comer, J.; Perkins, L. Resistance of the soil microbial community to land-surface disturbances of high-intensity winter grazing and wildfire. J. Environ. Manag. 2021, 279, 111596. [Google Scholar] [CrossRef]
- Rogiers, T.; Claesen, J.; Van Gompel, A.; Vanhoudt, N.; Mysara, M.; Williamson, A.; Leys, N.; Van Houdt, R.; Boon, N.; Mijnendonckx, K. Soil microbial community structure and functionality changes in response to long-term metal and radionuclide pollution. Environ. Microbiol. 2021, 23, 1670–1683. [Google Scholar] [CrossRef]
- Scholz, M.B.; Lo, C.-C.; Chain, P.S.G. Next generation sequencing and bioinformatic bottlenecks: The current state of metagenomic data analysis. Curr. Opin. Biotechnol. 2012, 23, 9–15. [Google Scholar] [CrossRef]
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Front. Plant. Sci. 2014, 5, 209. [Google Scholar] [CrossRef] [Green Version]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell. 2015, 58, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.H.; Liao, Y.C. Accurate binning of metagenomic contigs via automated clustering sequences using information of genomic signatures and marker genes. Sci. Rep. 2016, 6, 24175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A review of methods and databases for metagenomic classification and assembly. Brief. Bioinform. 2019, 20, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Méndez-García, C.; Bargiela, R.; Martínez-Martínez, M.; Ferrer, M. Chapter 2, Metagenomic protocols and strategies. In Metagenomics; Nagarajan, M., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 15–54. [Google Scholar] [CrossRef]
- Manzoni, S.; Schimel, J.P.; Porporato, A. Responses of soil microbial communities to water stress: Results from a meta-analysis. Ecology 2012, 93, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Myrold, D.D.; Zeglin, L.H.; Jansson, J.K. The Potential of Metagenomic Approaches for understanding soil microbial processes. Soil. Sci. Soc. Am. J. 2013, 78, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sedlar, K.; Kupkova, K.; Provaznik, I. Bioinformatics strategies for taxonomy independent binning and visualization of sequences in shotgun metagenomics. Comput. Struct. Biotechnol. J. 2017, 15, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, S.; Xu, Z.W.; Yan, Q.Y.; Li, X.B.; van Nostrand, J.D.; He, Z.; Yao, F.; Han, X.; Zhou, J.; et al. Responses of soil microbial functional genes to global changes are indirectly influenced by aboveground plant biomass variation. Soil. Biol. Biochem. 2017, 104, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Nunan, N.; Hirsch, P.R.; Sun, B.; Zhou, J.; Liang, Y. Theory of microbial coexistence in promoting soil–plant ecosystem health. Biol. Fert. Soils 2021, 57, 897–911. [Google Scholar] [CrossRef]
- Wang, Y.; Hong, Y.; Tian, Y.; Tian, G.; Zhang, J.; Wu, H.; Bai, Y.; Qian, J. Changes in bacterial community composition and soil properties altered the response of soil respiration to rain addition in desert biological soil crusts. Geoderma 2022, 409, 115635. [Google Scholar] [CrossRef]
- Peng, M.; Zi, X.; Wang, Q. Bacterial community diversity of oil contaminated soils assessed by high throughput sequencing of 16S rRNA genes. Int. J. Environ. Res. Public Health 2015, 12, 12002–12015. [Google Scholar] [CrossRef] [Green Version]
- Na, X.; Yu, H.; Wang, P.; Zhu, W.; Niu, Y.; Huang, J. Vegetation biomass and soil moisture coregulate bacterial community succession under altered precipitation regimes in a desert steppe in northwestern China. Soil. Biol. Biochem. 2019, 136, 107520. [Google Scholar] [CrossRef]
- Rutgers, M.; Wouterse, M.; Drost, S.M.; Breure, A.M.; Mulder, C.; Stone, D.; Winding, A.; Bloem, J. Monitoring soil bacteria with community-level physiological profiles using BiologTM ECO-plates in the Netherlands and Europe. Appl. Soil. Ecol. 2016, 97, 23–35. [Google Scholar] [CrossRef]
- Gałazka, A.; Grzadziel, J.; Gałazka, R.; Ukalska-Jaruga, A.; Strzelecka, J.; Smreczak, B. Genetic and functional diversity of bacterial microbiome in soils with long term impacts of petroleum hydrocarbons. Front. Microbiol. 2018, 9, 1923. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Zhang, Q.; Zhu, H.; Reich, P.B.; Banerjee, S.; van der Heijden, M.G.; Sadowsky, M.J.; Ishii, S.; Jia, X.; Shao, M.; et al. Erosion reduces soil microbial diversity, network complexity and multifunctionality. ISME J. 2021, 15, 2474–2489. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.environment.nsw.gov.au/topics/land-and-soil/soil-degradation/soil-biodiversi-ty#:~:text=Soil%20biodiversity%20is%20the%20variety,up%20to%206%20billion%20microorganisms (accessed on 30 June 2022).
- Available online: http://csbi.org.uk/our-work/tools-guidance/ (accessed on 30 June 2022).
- Malla, M.A.; Dubey, A.; Yadav, S.; Kumar, A.; Hashem, A.; Abd Allah, E.F. Understanding and designing the strategies for the microbe-mediated remediation of environmental contaminants using omics approaches. Front. Microbiol. 2018, 9, 1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichler, M.; Coskun, Ö.K.; Ortega-Arbulú, A.S.; Conci, N.; Wörheide, G.; Vargas, S.; Orsi, W.D. A 16S rRNA gene sequencing and analysis protocol for the Illumina MiniSeq platform. MicrobiologyOpen 2018, 7, e611. [Google Scholar] [CrossRef]
- Carbonetto, B.; Rascovan, N.; Alvarez, R.; Mentaberry, A.; Vazquez, M.P. Structure, composition and metagenomic profile of soil microbiomes associated to agricultural land use and tillage systems in Argentine Pampas. PLoS ONE 2014, 9, e99949. [Google Scholar] [CrossRef]
- Liu, Q.; Tang, J.; Gao, K.; Gurav, R.; Giesy, J.P. Aerobic degradation of crude oil by microorganisms in soils from four geographic regions of China. Sci. Rep. 2017, 7, 14856. [Google Scholar] [CrossRef] [Green Version]
- Klimek, B.; Sitarz, A.; Choczyński, M.; Niklińska, M. The effects of heavy metals and total petroleum hydrocarbons on soil bacterial activity and functional diversity in the Upper Silesia Industrial Region (Poland). Water Air Soil. Pollut. 2016, 227, 265. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, P.S.; Mishra, S.K.; Misra, S.; Dixit, V.K.; Pandey, S.; Khare, P.; Khan, M.H.; Dwivedi, S.; Lehri, A. Evaluation of fertility indicators associated with arsenic-contaminated paddy fields soil. Int. J. Environ. Sci. Technol. 2018, 15, 2447–2458. [Google Scholar] [CrossRef]
- Ma, S.; Qiao, L.; Liu, X.; Zhang, S.; Zhang, L.; Qiu, Z.; Yu, C. Microbial community succession in soils under long-term heavy metal stress from community diversity-structure to KEGG function pathways. Environ. Res. 2022, 214, 113822. [Google Scholar] [CrossRef]
- Deng, L.; Zeng, G.; Fan, C.; Lu, L.; Chen, X.; Chen, M.; Wu, H.; He, X.; He, Y. Response of rhizosphere microbial community structure and diversity to heavy metal co-pollution in arable soil. Appl. Microbiol. Biotechnol. 2015, 99, 8259–8269. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.Z.; Rafatullah, M. Recent advances in soil microbial fuel cells for soil contaminants remediation. Chemosphere 2021, 272, 129691. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Datta, S.; Chattopadhyay, D.; Sarkar, P. Arsenic accumulating and transforming bacteria isolated from contaminated soil for potential use in bioremediation. J. Environ. Sci. Health 2011, 46, 1736–1747. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Nasir, M.; Lv, J.; Dai, Y.; Gao, J. Understanding the variation of microbial community in heavy metals contaminated soil using high throughput sequencing. Ecotoxicol. Environ. Saf. 2017, 144, 300–306. [Google Scholar] [CrossRef]
- Hu, X.; Liu, X.; Qiao, L.; Zhang, S.; Su, K.; Qiu, Z.; Li, X.; Zhao, Q.; Yu, C. Study on the spatial distribution of ureolytic microorganisms in farmland soil around tailings with different heavy metal pollution. Sci. Total. Environ. 2021, 775, 144946. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, T.; Cheng, J.; Wei, G.; Lin, Y. Above- and belowground biodiversity drives soil multifunctionality along a long-term grassland restoration chronosequence. Sci. Total. Environ. 2021, 772, 145010. [Google Scholar] [CrossRef]
- Covino, S.; Fabianová, T.; Křesinová, Z.; Čvančarová, M.; Burianová, E.; Filipová, A.; Vořísková, J.; Baldrian, P.; Cajthaml, T. Polycyclic aromatic hydrocarbons degradation and microbial community shifts during co-composting of creosote-treated wood. J. Hazard. Mater. 2016, 301, 17–26. [Google Scholar] [CrossRef]
- Jia, T.; Wang, Y.; Chai, B. Bacterial community characteristics and enzyme activities in Bothriochloa ischaemum litter over progressive phytoremediation years in a copper tailings dam. Front. Microbiol. 2020, 11, 565806. [Google Scholar] [CrossRef]
- Máthé, I.; Benedek, T.; Táncsics, A.; Palatinszky, M.; Lányi, S.; Márialigeti, K. Diversity, activity, antibiotic and heavy metal resistance of bacteria from petroleum hydrocarbon contaminated soils located in Harghita County (Romania). Int. Biodeter Biodegrad. 2012, 73, 41–49. [Google Scholar] [CrossRef]
- Alisi, C.; Musella, R.; Tasso, F.; Ubaldi, C.; Manzo, S.; Cremisini, C.; Sprocati, A.R. Bioremediation of diesel oil in a co-contaminated soil by bioaugmentation with a microbial formula tailored with native strains selected for heavy metals resistance. Sci. Tot. Environ. 2009, 407, 3024–3032. [Google Scholar] [CrossRef]
- Sprocati, A.R.; Alisi, C.; Tasso, F.; Marconi, P.; Sciullo, A.; Pinto, V.; Chiavarini, S.; Ubaldi, C.; Cremisini, C. Effectiveness of a microbial formula, as a bioaugmentation agent, tailored for bioremediation of diesel oil and heavy metal co-contaminated soil. Process. Biochem. 2012, 47, 1649–1655. [Google Scholar] [CrossRef]
- Zhong, Y.; Luan, T.; Lin, L.; Liu, H.; Tam, N.F.Y. Production of metabolites in the biodegradation of phenanthrene, fluoranthene and pyrene by the mixed culture of Mycobacterium sp. and Sphingomonas sp. Bioresource Technol. 2011, 102, 2965–2972. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Wu, M.; Liu, H.; Xu, Y.; Liu, Z. Effect of petroleum hydrocarbon pollution levels on the soil microecosystem and ecological function. Environ. Pollut. 2022, 293, 118511. [Google Scholar] [CrossRef]
- Sutton, N.B.; Maphosa, F.; Morillo, J.; Abu Al-Soud, W.; Langenhoff, A.A.M.; Grotenhuis, T. Impact of long-term diesel contamination on soil microbial community structure. Appl. Environ. Microbiol. 2013, 79, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Naether, A.; Foesel, B.U.; Naegele, V.; Wüst, P.K.; Weinert, J.; Bonkowski, M. Environmental factors affect actinobacterial communities below the subgroup level in grassland and forest soils. Appl. Environ. Microbiol. 2012, 78, 7398–7406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegner, C.E.; Liesack, W. Microbial community dynamics during the early stages of plant polymer breakdown in paddy soil. Environ. Microbiol. 2016, 18, 2825–2842. [Google Scholar] [CrossRef]
- Msaddak, A.; Durán, D.; Rejili, M.; Mars, M.; Ruiz-Argüeso, T.; Imperial, J.; Palacios, J.; Rey, L. Diverse bacteria affiliated with the genera Microvirga, Phyllobacterium, and Bradyrhizobium modulate Lupinus micranthus growing in soils of Northern Tunisia. Appl. Environ. Microbiol. 2017, 2, 83. [Google Scholar]
- Sitte, J.; Akob, D.M.; Kaufmann, C.; Finster, K.; Banerjee, D.; Burkhardt, E.M.; Kostka, J.E.; Scheinost, A.C.; Büchel, G.; Küsel, K. Microbial links between sulfate reduction and metal retention in uranium- and heavy metal-contaminated soil. Appl. Environ. Microbiol. 2010, 76, 3143–3152. [Google Scholar] [CrossRef] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonomo, M.G.; Scrano, L.; Mang, S.M.; Scalese, B.E.; Bufo, S.A.; Modley, L.-A.; Buongarzone, E.; Salzano, G. Changes in the Bacterial Community Composition of Cultivated Soil after Digging up Operations for Laying a Pipeline. Agriculture 2023, 13, 1189. https://doi.org/10.3390/agriculture13061189
Bonomo MG, Scrano L, Mang SM, Scalese BE, Bufo SA, Modley L-A, Buongarzone E, Salzano G. Changes in the Bacterial Community Composition of Cultivated Soil after Digging up Operations for Laying a Pipeline. Agriculture. 2023; 13(6):1189. https://doi.org/10.3390/agriculture13061189
Chicago/Turabian StyleBonomo, Maria Grazia, Laura Scrano, Stefania Mirela Mang, Barbara Emanuela Scalese, Sabino Aurelio Bufo, Lee-Ann Modley, Euro Buongarzone, and Giovanni Salzano. 2023. "Changes in the Bacterial Community Composition of Cultivated Soil after Digging up Operations for Laying a Pipeline" Agriculture 13, no. 6: 1189. https://doi.org/10.3390/agriculture13061189