Understanding the Relationship between Glutathione, TGF-β, and Vitamin D in Combating Mycobacterium tuberculosis Infections

 ,
,  ,
,  and
and 
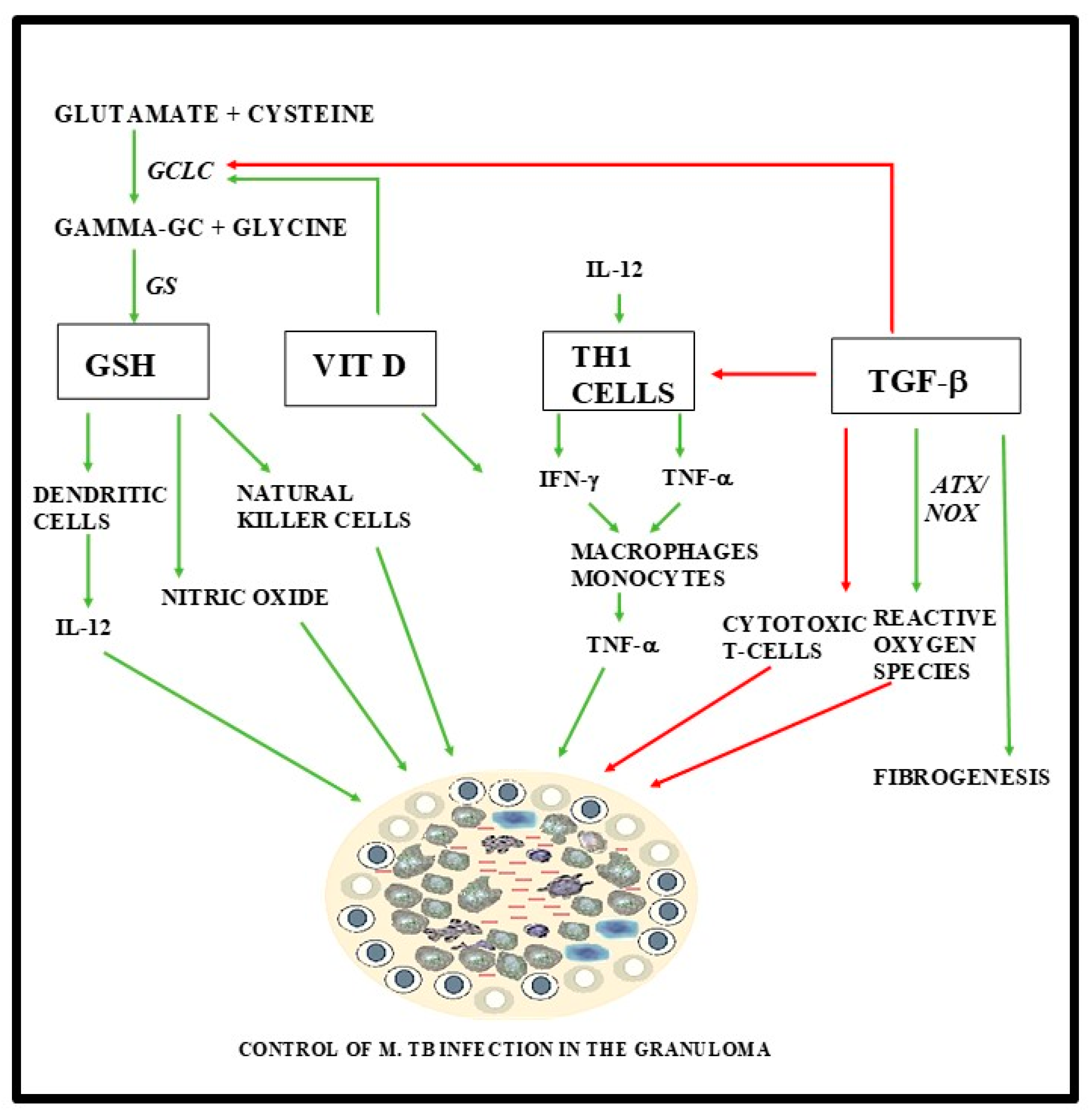{kind=link}
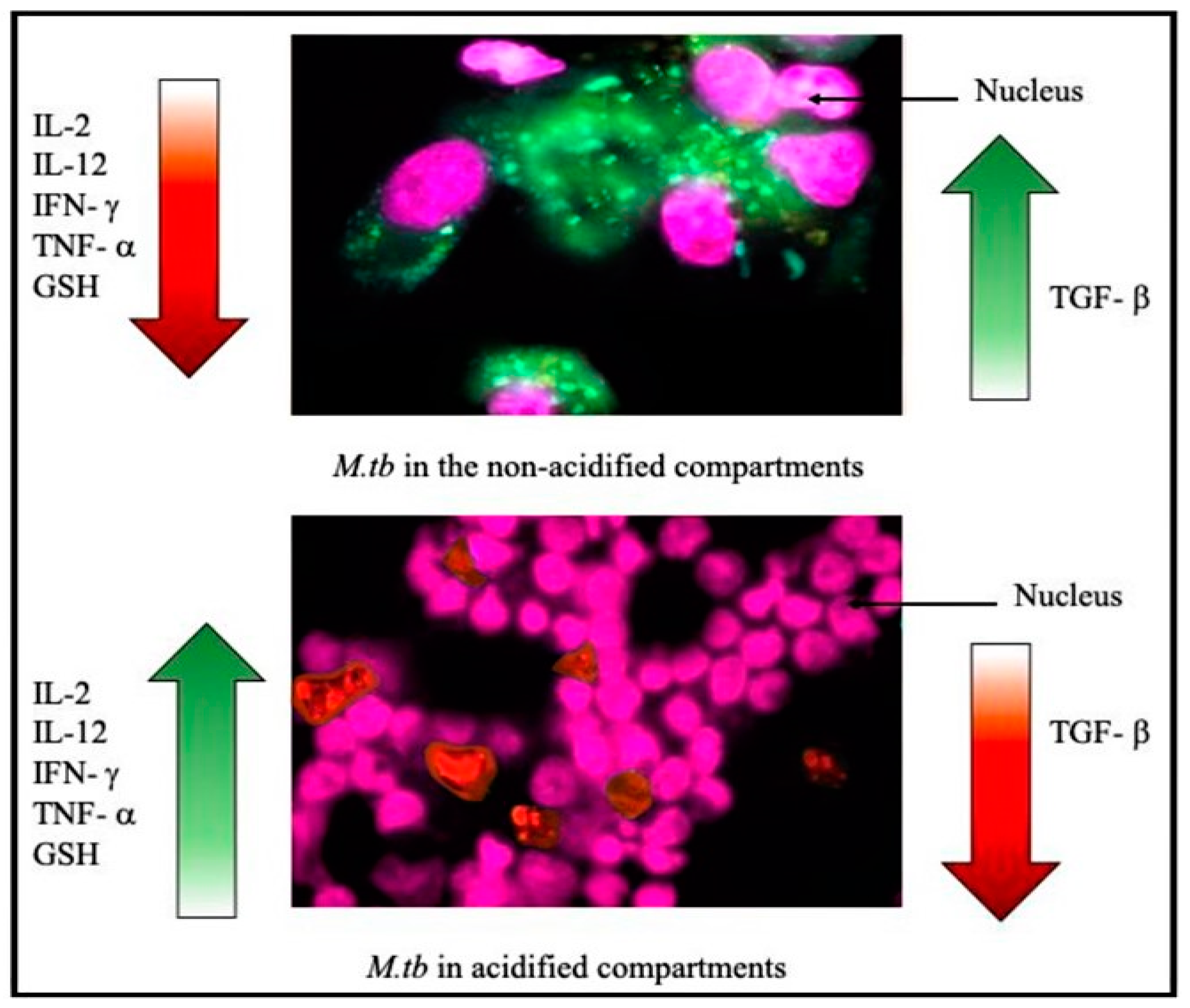{kind=link}
Abstract
:1. Mycobacterium tuberculosis and the Host Immune Responses
1.1. Mycobacterium tuberculosis Infection
1.2. Cytokines Profile in the Immune Response to M. tb
1.3. The Role of Glutathione, TGF-B, and Vitamin D in the Immune Response
1.4. Clinical Significance
2. The Effects of Glutathione on the Immune System
2.1. The Role of GSH in Maintaining Cellular Redox Homeostasis
2.2. Immunomodulatory Effects of GSH
3. TGF-β in the Immune Responses
3.1. Activation of TGF-β
3.2. TGF-β and GSH
3.3. TGF-β and Tuberculosis
4. Vitamin D, TB, and the Immune Responses
4.1. Cellular and Immunological Response to Vitamin D
4.2. Vitamin D and TB
5. HIV and TB
GSH Supplementation in Increasing Cytokine Production in HIV Patients
6. Type-2 Diabetes Mellitus and TB
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019; License: CCBY-NC-SA3.0IGO. 2019.
- Pease, C.; Hutton, B.; Yazdi, F.; Wolfe, D.; Hamel, C.; Quach, P.; Skidmore, B.; Moher, D.; Alvarez, G.G. Efficacy and completion rates of rifapentine and isoniazid (3HP) compared to other treatment regimens for latent tuberculosis infection: A systematic review with network meta-analyses. BMC Infect. Dis. 2017, 17. [Google Scholar] [CrossRef]
- Lowe, D.M.; Redford, P.S.; Wilkinson, R.J.; O’Garra, A.; Martineau, A.R. Neutrophils in tuberculosis: Friend or foe? Trends Immunol. 2012, 33, 14–25. [Google Scholar] [CrossRef]
- Basile, J.I.; Kviatcovsky, D.; Romero, M.M.; Balboa, L.; Monteserin, J.; Ritacco, V.; Lopez, B.; García, C.S.; García, A.; Vescovo, M.; et al. Mycobacterium tuberculosis multi-drug-resistant strain M induces IL-17+ IFNγ- CD4+ T cell expansion through an IL-23 and TGF-β-dependent mechanism in patients with MDR-TB tuberculosis. Clin. Exp. Immunol. 2017, 187, 160–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagán, A.J.; Ramakrishnan, L. The Formation and Function of Granulomas. Annu. Rev. Immunol. 2018, 36, 639–665. [Google Scholar] [CrossRef] [PubMed]
- Cronan, M.; Beerman, R.; Rosenberg, A.; Saelens, J.; Johnson, M.; Oehlers, S.; Sisk, D.M.; Jurcic, K.L.; Medvitz, S.N.A.; Miller, S.E.; et al. Macrophage Epithelial Reprogramming Underlies Mycobacterial Granuloma Formation and Promotes Infection. Immunity 2016, 45, 861–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shkurupiy, V.A.; Kim, L.B.; Potapova, O.V.; Cherdantseva, L.A.; Putyatina, A.N.; Nikonova, I.K. Fibrogenesis in Granulomas and Lung Interstitium in Tuberculous Inflammation in Mice. Bull. Exp. Biol. Med. 2014, 156, 731–735. [Google Scholar] [CrossRef]
- Natarajan, K.; Kundu, M.; Sharma, P.; Basu, J. Innate immune responses to M. tuberculosis infection. Tuberculosis 2011, 91, 427–431. [Google Scholar] [CrossRef]
- Vergne, I.; Fratti, R.A.; Hill, P.J.; Chua, J.; Belisle, J.; Deretic, V. Mycobacterium tuberculosis phagosome maturation arrest: Mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Mol. Biol. Cell 2004, 15, 751–760. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, J.S.; Adetifa, I.M.; Hill, P.C.; Adegbola, R.A.; Ota, M.O. Pattern and diversity of cytokine production differentiates between Mycobacterium tuberculosis infection and disease. Eur. J. Immunol. 2009, 39, 723–729. [Google Scholar] [CrossRef]
- Boom, W.H.; Canaday, D.H.; Fulton, S.A.; Gehring, A.J.; Rojas, R.E.; Torres, M. Human immunity to M. tuberculosis: T cell subsets and antigen processing. Tuberculosis 2003, 83, 98–106. [Google Scholar] [CrossRef]
- Verhasselt, V.; Berghe, W.V.; Vanderheyde, N.; Willems, F.; Haegeman, G.; Goldman, M. N-acetyl-l-cysteine inhibits primary human T cell responses at the dendritic cell level: Association with NF-kappaB inhibition. J. Immunol. 1999, 162, 2569–2574. [Google Scholar] [PubMed]
- Kaufmann, S.H.E. Protection against tuberculosis: Cytokines, T cells, and macrophages. Ann. Rheum. Dis. 2002, 61, i54–i58. [Google Scholar] [CrossRef] [PubMed]
- Philips, J.A.; Ernst, J.D. Tuberculosis Pathogenesis and Immunity. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 353–384. [Google Scholar] [CrossRef] [PubMed]
- Warsinske, H.C.; Pienaar, E.; Linderman, J.J.; Mattila, J.T.; Kirschner, D.E. Deletion of TGF-β1 Increases Bacterial Clearance by Cytotoxic T Cells in a Tuberculosis Granuloma Model. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Shan, B.; Lasky, J.A. TGF-β: Titan of Lung Fibrogenesis. Curr. Enzym. Inhib. 2010, 6, 67–77. [Google Scholar] [CrossRef]
- Lagman, M.; Ly, J.; Saing, T.; Singh, M.K.; Tudela, E.V.; Morris, D.; Chi, P.; Ochoa, C.; Sathananthan, A.; Venketaraman, V. Investigating the Causes for Decreased Levels of Glutathione in Individuals with Type II Diabetes. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Harris, J. Autophagy and cytokines. Cytokine 2011, 56, 140–144. [Google Scholar] [CrossRef]
- Ghezzi, P. Role of glutathione in immunity and inflammation in the lung. Int. J. Gen. Med. 2011, 4, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Rahman, I.; Biswas, S.K.; Jimenez, L.A.; Torres, M.; Forman, H.J. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid. Redox Signal. 2005, 7, 42–59. [Google Scholar] [CrossRef]
- Teskey, G.; Cao, R.; Islamoglu, H.; Medina, A.; Prasad, C.; Prasad, R.; Sathananthan, A.; Fraix, M.; Subbian, S.; Zhong, L.; et al. The Synergistic Effects of the Glutathione Precursor, NAC and First-Line Antibiotics in the Granulomatous Response Against Mycobacterium tuberculosis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.; Ly, J.; Chi, P.T.; Daliva, J.; Nguyen, T.; Soofer, C.; Chen, Y.C.; Lagman, M.; Venketaraman, V. Glutathione synthesis is compromised in erythrocytes from individuals with HIV. Front. Pharmacol. 2014, 5, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutchmansingh, F.; Hsu, J.; Bennett, F.; Badaloo, A.; McFarlane-Anderson, N.; Gordon-Strachan, G.; Wright-Pascoe, R.; Jahoor, F.; Boyne, M. Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLoS ONE 2018, 13, e0198626. [Google Scholar] [CrossRef] [Green Version]
- Ferlita, S.; Yegiazaryan, A.; Noori, N.; Lal, G.; Nguyen, T.; To, K.; Venketaraman, V. Type 2 Diabetes Mellitus and Altered Immune System Leading to Susceptibility to Pathogens, Especially Mycobacterium tuberculosis. J. Clin. Med. 2019, 8, 2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricca, C.; Aillon, A.; Viano, M.; Bergandi, L.; Aldieri, E.; Silvagno, F. Vitamin D inhibits the epithelial-mesenchymal transition by a negative feedback regulation of TGF-β activity. J. Steroid Biochem. Mol. Biol. 2019, 187, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Airey, F.S. Vitamin D as a remedy for lupus vulgaris. Med. World 1946, 64, 807–810. [Google Scholar]
- World Health Organization. Tuberculosis and HIV. Available online: https://www.who.int/hiv/topics/tb/en/ (accessed on 8 July 2020).
- Bruchfeld, J.; Correia-Neves, M.; Källenius, G. Tuberculosis and HIV Coinfection: Table 1. Cold Spring Harb. Perspect. Med. 2015, 5, a017871. [Google Scholar] [CrossRef]
- Liu, R.M.; Pravia, K.G. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic. Biol. Med. 2010, 48, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, P.C. Cytoplasmic and Mitochondrial NADPH-Coupled Redox Systems in the Regulation of Aging. Nutrients 2019, 11, 504. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.S.; Chang, L.S.; Anderson, M.E.; Meister, A. Catalytic and Regulatory Properties of the Heavy Subunit of Rat Kidney y-Glutamylcysteine Synthetase. J. Biol. Chem. 1993, 268, 19675–19680. [Google Scholar]
- Meister, A. On the cycles of glutathione metabolism and transport. In Current Topics in Cellular Regulation; Academic Press: Chicago, IL, USA, 1981; Volume 18, pp. 21–58. [Google Scholar]
- Roum, J.H.; Buhl, R.; McElvany, N.G.; Borok, Z.; Crystal, R.G. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1993, 75, 2419–2424. [Google Scholar] [CrossRef]
- Tirouvanziam, R.; Conrad, C.K.; Bottiglieri, T.; Herzenberg, L.A.; Moss, R.B. High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4628–4633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeh, K.M.; Beier, J.; Haas, I.C.; Kornmann, O.; Micke, P.; Buhl, R. Glutathione deficiency of the lower respiratory tract in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2002, 19, 1119–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behr, J.; Degenkolb, B.; Krombach, F.; Vogelmeier, C. Intracellular glutathione and bronchoalveolar cells in fibrosing alveolitis: Effects of N-acetylcysteine. Eur. Respir. J. 2002, 19, 906–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behr, J.; Maier, K.; Degenkolb, B.; Krombach, F.; Vogelmeier, C. Antioxidative and clinical effects of high-dose N-acetylcysteine in fibrosing alveolitis. Adjunctive therapy to maintenance immunosuppression. Am. J. Respir. Crit. Care Med. 1997, 156, 1897–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borok, Z.; Grimes, G.J.; Buhl, R.; Bokser, A.; Hubbard, R.C.; Holroyd, K.; Roum, J.H.; Czerski, D.B.; Cantin, A.M.; Crystal, R.G. Effect of glutathione aerosol on oxidant-antioxidant imbalance in idiopathic pulmonary fibrosis. Lancet 1991, 338, 215–216. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hubbard, R.C.; Crystal, R.G. Glutathione deficiency in the epithelial lining fluid of the lower respiratory tract in idiopathic pulmonary fibrosis. Am. Rev. Respir. Dis. 1989, 139, 370–372. [Google Scholar] [CrossRef]
- Meyer, A.; Buhl, R.; Magnussen, H. The effect of oral N-acetylcysteine on lung glutathione levels in idiopathic pulmonary fibrosis. Eur. Respir. J. 1994, 7, 431–436. [Google Scholar] [CrossRef]
- Montaldo, C.; Cannas, E.; Ledda, M.; Rosetti, L.; Congiu, L.; Atzori, L. Bronchoalveolar glutathione and nitrite/nitrate in idiopathic pulmonary fibrosis and sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2002, 19, 54–58. [Google Scholar]
- Boots, A.W.; Drent, M.; Swennen, E.L.R.; Moonen, H.J.J.; Bast, A.; Haenen, G.R.M.M. Antioxidant status associated with inflammation in sarcoidosis: A potential role for antioxidants. Respir. Med. 2009, 103, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Fraternale, A.; Brundu, S.; Magnani, M. Glutathione and glutathione derivatives in immunotherapy. Biol. Chem. 2017, 398, 261–275. [Google Scholar] [CrossRef]
- Teskey, G.; Abrahem, R.; Cao, R.; Gyurjian, K.; Islamoglu, H.; Lucero, M.; Martineza, A.; Paredes, E.; Salaiz, O.; Robinson, B.; et al. Glutathione as a Marker for Human Disease. Adv. Clin. Chem. 2018, 87, 141–159. [Google Scholar] [CrossRef] [PubMed]
- Ly, J.; Lagman, M.; Saing, T.; Singh, M.K.; Tudela, E.V.; Morris, D.; Anderson, J.; Daliva, J.; Ochoa, C.; Patel, N.; et al. Liposomal Glutathione Supplementation Restores TH1 Cytokine Response to Mycobacterium tuberculosis Infection in HIV-Infected Individuals. J. Interferon Cytokine Res. 2015, 35, 875–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, J.J. Glutathione depletion is associated with augmenting a proinflammatory signal: Evidence for an antioxidant/pro-oxidant mechanism regulating cytokines in the alveolar epithelium. Cytokines Cell Mol. Ther. 2000, 6, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Bailey, C.; Cahatol, I.; Dodge, L.; Yim, J.; Kassissa, C.; Luong, J.; Kasko, S.; Pandya, S.; Venketaraman, V. Mechanisms of Control of Mycobacterium tuberculosis by NK Cells: Role of Glutathione. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.; Nguyen, T.; Kim, J.; Kassissa, C.; Khurasany, M.; Luong, J.; Kasko, S.; Pandya, S.; Chu, M.; Chi, P.-T.; et al. An Elucidation of Neutrophil Functions against Mycobacterium tuberculosis Infection. Clin. Dev. Immunol. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.; Gonzalez, B.; Khurasany, M.; Kassissa, C.; Luong, J.; Kasko, S.; Pandya, S.; Chu, M.; Chi, P.; Bui, S.; et al. Characterization of Dendritic Cell and Regulatory T Cell Functions against Mycobacterium tuberculosis Infection. BioMed Res. Int. 2013, 2013, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Nikitovic, D.; Holmgren, A. S-nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. J. Biol. Chem. 1996, 271, 19180–19185. [Google Scholar] [CrossRef] [Green Version]
- Venketaraman, V.; Dayaram, Y.K.; Talaue, M.T.; Connell, N.D. Glutathione and Nitrosoglutathione in Macrophage Defense against Mycobacterium tuberculosis. Infect. Immun. 2005, 73, 1886–1889. [Google Scholar] [CrossRef] [Green Version]
- Millman, A.C.; Salman, M.; Dayaram, Y.K.; Connell, N.D.; Venketaraman, V. Natural Killer Cells, Glutathione, Cytokines, and Innate Immunity AgainstMycobacterium tuberculosis. J. Interferon Cytokine Res. 2008, 28, 153–165. [Google Scholar] [CrossRef]
- Valdivia, A.; Ly, J.; Gonzalez, L.; Hussain, P.; Saing, T.; Islamoglu, H.; Pearce, D.; Ochoa, C.; Venketaraman, V. Restoring Cytokine Balance in HIV-Positive Individuals with Low CD4 T Cell Counts. AIDS Res. Hum. Retrovir. 2017, 33, 905–918. [Google Scholar] [CrossRef]
- Garg, S.K.; Yan, Z.; Vitvitsky, V.; Banerjee, R. Differential Dependence on Cysteine from Transsulfuration versus Transport During T Cell Activation. Antioxid. Redox Signal. 2011, 15, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Kamide, Y.; Utsugi, M.; Dobashi, K.; Ono, A.; Ishizuka, T.; Hisada, T.; Koga, Y.; Uno, K.; Hamuro, J.; Mori, K. Intracellular glutathione redox status in human dendritic cells regulates IL-27 production and T-cell polarization. Allergy 2011, 66, 1183–1192. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef] [Green Version]
- Franklin, C.C.; Rosenfeld-Franklin, M.E.; White, C.; Kavanagh, T.J.; Fausto, N. TGFβ1-induced suppression of glutathione antioxidant defenses in hepatocytes: Caspase-dependent posttranslational and caspase-independent transcriptional regulatory mechanisms. FASEB J. 2003, 17, 1–23. [Google Scholar] [CrossRef]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Zi, Z.; Chapnick, D.A.; Liu, X. Dynamics of TGF-β/Smad signaling. Febs. Lett. 2012, 586, 1921–1928. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Jiang, Y.; Wang, Q.; Ma, X.; Xiao, Z.; Zuo, W.; Fang, X.; Cheng, Y.-G. Single-molecule imaging reveals transforming growth factor-beta-induced type II receptor dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 15679–15683. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Shenvi, S.V.; Smith, E.; Hagen, T.M. Identification of age-specific Nrf2 binding to a novel antioxidant response element locus in the Gclc promoter: A compensatory means for the loss of glutathione synthetic capacity in the aging rat liver? Aging Cell 2012, 11, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharm. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yu, S.; Liu, T.; Kim, J.-H.; Blank, V.; Li, H.; Kong, A.-N.T. Heterodimerization with small Maf proteins enhances nuclear retention of Nrf2 via masking the NESzip motif. Biochim. Biophys. Acta 2008, 1783, 1847–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa-Diez, C.; Fierro-Fernández, M.; Sánchez-Gómez, F.; Rodríguez-Pascual, F.; Alique, M.; Ruiz-Ortega, M.; Beraza, N.; Martínez-Chantar, M.L.; Fernández-Hernando, C.; Lamas, S. Targeting of Gamma-Glutamyl-Cysteine Ligase by miR-433 Reduces Glutathione Biosynthesis and Promotes TGF-β-Dependent Fibrogenesis. Antioxid. Redox Signal. 2015, 23, 1092–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aung, H.; Wu, M.; Johnson, J.; Hirsch, C.; Toossi, Z. Bioactivation of Latent Transforming Growth Factor beta1 by Mycobacterium tuberculosis in Human Mononuclear Phagocytes. Scand. J. Immunol. 2005, 61, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Bonecini-Almeida, M.G.; Ho, J.L.; Boéchat, N.; Huard, R.C.; Chitale, S.; Doo, H.; Geng, J.; Rego, L.; Lazzarini, L.C.; Kritski, A.L.; et al. Down-modulation of lung immune responses by interleukin-10 and transforming growth factor beta (TGF-beta) and analysis of TGF-beta receptors I and II in active tuberculosis. Infect. Immun. 2004, 72, 2628–2634. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Liu, G.S.; Dusting, G.J.; Chan, E.C. NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic responses. Redox Biol. 2014, 2, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.; Xie, Y.; Abel, P.W.; Huang, Y.; Ma, Q.; Li, L.; Hao, J.; Wolff, D.W.; Wei, T.; Tu, Y. Transforming growth factor (TGF)-β1-induced miR-133a inhibits myofibroblast differentiation and pulmonary fibrosis. Cell Death Dis. 2019, 10, 670. [Google Scholar] [CrossRef]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2016, 64, 157–167. [Google Scholar] [CrossRef]
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Dijke, P.T. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18, 2157. [Google Scholar] [CrossRef] [Green Version]
- Difazio, R.M.; Mattila, J.T.; Klein, E.C.; Cirrincione, L.R.; Howard, M.; Wong, E.A.; Flynn, J.L. Active transforming growth factor-β is associated with phenotypic changes in granulomas after drug treatment in pulmonary tuberculosis. Fibrogenesis Tissue Repair 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Bikle, D.D. Extrarenal Synthesis of 1,25-Dihydroxyvitamin D and Its Health Implications. Vitamin D 2010, 277–295. [Google Scholar] [CrossRef]
- Toossi, Z.; Ellner, J.J. The Role of TGFβ in the Pathogenesis of Human Tuberculosis. Clin. Immunol. Immunopathol. 1998, 87, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.L.; Dartois, V.; Johnston, P.J.; Janssen, C.; Via, L.; Goodwin, M.B.; Klein, E.; Barry, C.E.; Flynn, J.L. Metronidazole prevents reactivation of latent Mycobacterium tuberculosis infection in macaques. Proc. Natl. Acad. Sci. USA 2012, 109, 14188–14193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghuvanshi, S.; Sharma, P.; Singh, S.; Van Kaer, L.; Das, G. Mycobacterium tuberculosis evades host immunity by recruiting mesenchymal stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 21653–21658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dis, E.; Sogi, K.M.; Rae, C.S.; Sivick, K.E.; Surh, N.H.; Leong, M.L.; Kanne, D.B.; Metchette, K.; Leong, J.J.; Bruml, J.R.; et al. STING-Activating Adjuvants Elicit a Th17 Immune Response and Protect against Mycobacterium tuberculosis Infection. Cell Rep. 2018, 23, 1435–1447. [Google Scholar] [CrossRef]
- Wu, M.; Aung, H.; Hirsch, C.S.; Toossi, Z. Inhibition of Mycobacterium tuberculosis-induced signalling by transforming growth factor-β in human mononuclear phagocytes. Scand. J. Immunol. 2012, 75, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Evans, R.M. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835. [Google Scholar] [CrossRef] [Green Version]
- Christakos, S.; Raval-Pandya, M.; Wernyj, R.P.; Yang, W. Genomic mechanisms involved in the pleiotropic actions of 1,25-dihydroxyvitamin D3. Biochem. J. 1996, 316, 361–371. [Google Scholar] [CrossRef]
- Bouillon, R.; Okamura, W.H.; Norman, A.W. Structure-function relationships in the vitamin D endocrine system. Endocr. Rev. 1995, 16, 200–257. [Google Scholar]
- James, S.; Williams, M.; Newland, A.; Colston, K. Leukemia cell differentiation: Cellular and molecular interactions of retinoids and vitamin D. Gen. Pharmacol. Vasc. Syst. 1999, 32, 143–154. [Google Scholar] [CrossRef]
- Wang, T.T.; Nestel, F.P.; Bourdeau, V.; Nagai, Y.; Wang, Q.; Liao, J.; Tavera-Mendoza, L.; Lin, R.; Hanrahan, J.W.; Mader, S.; et al. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J. Immunol. 2004, 173, 2909–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gombart, A.F.; Borregaard, N.; Koeffler, H.P. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J. 2005, 19, 1067–1077. [Google Scholar] [CrossRef] [Green Version]
- Gombart, A.F.; O’Kelly, J.; Saito, T.; Koeffler, H.P. Regulation of the CAMPgene by 1,25(OH)2D3 in various tissues. J. Steroid Biochem. Mol. Biol. 2007, 103, 552–557. [Google Scholar] [CrossRef]
- Liu, P.T.; Schenk, M.; Walker, V.P.; Dempsey, P.W.; Kanchanapoomi, M.; Wheelwright, M.; Vazirnia, A.; Zhang, X.; Steinmeyer, A.; Zügel, U.; et al. Convergence of IL-1β and VDR activation pathways in human TLR2/1-induced antimicrobial responses. PLoS ONE 2009, 4, e5810. [Google Scholar] [CrossRef] [Green Version]
- Weber, G.; Heilborn, J.D.; Jimenez, C.I.C.; Hammarsjo, A.; Torma, H.; Stahle, M. Vitamin D induces the antimicrobial protein hCAP18 in human skin. J. Investig. Derm. 2005, 124, 1080–1082. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Kamen, D.L. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef]
- Schauber, J.; Dorschner, R.A.; Coda, A.B.; Büchau, A.S.; Liu, P.T.; Kiken, D.; Zügel, U. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D—Dependent mechanism. J. Clin. Investig. 2007, 117, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Fabri, M.; Stenger, S.; Shin, D.M.; Yuk, J.M.; Liu, P.T.; Realegeno, S.; Lee, H.M.; Krutzik, S.R.; Schenk, M.; Sieling, P.A.; et al. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci. Transl. Med. 2011, 3, 104ra102. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.K.; Micinski, D. Vitamin D upregulates glutamate cysteine ligase and glutathione reductase, and GSH formation, and decreases ROS and MCP-1 and IL-8 secretion in high-glucose exposed U937 monocytes. Biochem. Biophys. Res. Commun. 2013, 437, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Grant, W.B.; Lahore, H.; McDonnell, S.L.; Baggerly, C.A.; French, C.B.; Aliano, J.L.; Bhattoa, H.P. Evidence that Vitamin D Supplementation Could Reduce Risk of Influenza and COVID-19 Infections and Deaths. Nutrients 2020, 12, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, R.J.; Llewelyn, M.; Toossi, Z.; Patel, P.; Pasvol, G.; Lalvani, A.; Wright, D.; Latif, M.; Davidson, R.N. Influence of vitamin D deficiency and vitamin D receptor polymorphisms on tuberculosis among Gujarati Asians in west London: A case-control study. Lancet 2000, 355, 618–621. [Google Scholar] [CrossRef]
- Jolliffe, D.A.; Ganmaa, D.; Wejse, C.; Raqib, R.; Haq, M.A.; Salahuddin, N.; Daley, P.K.; Ralph, A.P.; Ziegler, T.R.; Martineau, A.R. Adjunctive vitamin D in tuberculosis treatment: Meta-analysis of individual participant data. Eur. Respir. J. 2019, 53, 1802003. [Google Scholar] [CrossRef] [Green Version]
- Yamshchikov, A.; Desai, N.; Blumberg, H.; Ziegler, T.; Tangpricha, V. Vitamin D for Treatment and Prevention of Infectious Diseases: A Systematic Review of Randomized Controlled Trials. Endocr. Pract. 2009, 15, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Holick, M.F.; Biancuzzo, R.M.; Chen, T.C.; Klein, E.K.; Young, A.; Bibuld, D.; Tannenbaum, A.D. Vitamin D2 is as effective as vitamin D3 in maintaining circulating concentrations of 25-hydroxyvitamin D. J. Clin. Endocrinol. Metab. 2008, 93, 677–681. [Google Scholar] [CrossRef]
- Tenforde, M.W.; Yadav, A.; Dowdy, D.W.; Gupte, N.; Shivakoti, R.; Yang, W.T.; Mwelase, N.; Kanyama, C.; Pillay, S.; Samaneka, W.; et al. Vitamin A and D Deficiencies Associated With Incident Tuberculosis in HIV-Infected Patients Initiating Antiretroviral Therapy in Multinational Case-Cohort Study. J. Acquir. Immune Defic. Syndr. 1999, 75, e71–e79. [Google Scholar] [CrossRef]
- Ku, N.S.; Oh, J.K.; Shin, S.Y.; Kim, S.B.; Kim, H.; Jeong, S.J.; Han, S.H.; Song, Y.G.; Kim, J.M.; Choi, J.Y. Effects of Tuberculosis on the Kinetics of CD4+ T Cell Count Among HIV-Infected Patients Who Initiated Antiretroviral Therapy Early After Tuberculosis Treatment. AIDS Res. Hum. Retrovir. 2013, 29, 226–230. [Google Scholar] [CrossRef]
- Lin, P.L.; Rutledge, T.; MGreen, A.; Bigbee, M.; Fuhrman, C.; Klein, E.; Flynn, J.L. CD4 T Cell Depletion Exacerbates Acute Mycobacterium Tuberculosis While Reactivation of Latent Infection Is Dependent on Severity of Tissue Depletion In Cynomolgus Macaques. AIDS Res. Hum. Retrovir. 2012, 28, 1693–1702. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Diabetes Facts (Infographics); WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Oates, P. Aldose Reductase, Still a Compelling Target for Diabetic Neuropathy. Curr. Drug Targets 2008, 9, 14–36. [Google Scholar] [CrossRef]
- Morré, D.M.; Lenaz, G.; Morré, D.J. Surface oxidase and oxidative stress propagation in aging. J. Exp. Biol. 2000, 203, 1513–1521. [Google Scholar]
- Cheng, H.-M.; González, R. The effect of high glucose and oxidative stress on lens metabolism, aldose reductase, and senile cataractogenesis. Metabolism 1986, 35, 10–14. [Google Scholar] [CrossRef]
- Gong, J.H.; Zhang, M.; Modlin, R.L.; Linsley, P.S.; Iyer, D.; Lin, Y.; Barnes, P.F. Interleukin-10 downregulates Mycobacterium tuberculosis-induced Th1 responses and CTLA-4 expression. Infect. Immun. 1996, 64, 913–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakin, A.V.; Stourman, N.V.; Sekhar, K.R.; Rinehart, C.; Yan, X.; Meredith, M.J.; Arteaga, C.L.; Freeman, M.L. Smad3-ATF3 signaling mediates TGF-β suppression of genes encoding Phase II detoxifying proteins. Free Radic. Biol. Med. 2005, 38, 375–387. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, M.; Vaughn, C.; Sasaninia, K.; Yeh, C.; Mehta, D.; Khieran, I.; Venketaraman, V. Understanding the Relationship between Glutathione, TGF-β, and Vitamin D in Combating Mycobacterium tuberculosis Infections. J. Clin. Med. 2020, 9, 2757. https://doi.org/10.3390/jcm9092757
Singh M, Vaughn C, Sasaninia K, Yeh C, Mehta D, Khieran I, Venketaraman V. Understanding the Relationship between Glutathione, TGF-β, and Vitamin D in Combating Mycobacterium tuberculosis Infections. Journal of Clinical Medicine. 2020; 9(9):2757. https://doi.org/10.3390/jcm9092757
Chicago/Turabian StyleSingh, Mohkam, Charles Vaughn, Kayvan Sasaninia, Christopher Yeh, Devanshi Mehta, Ibrahim Khieran, and Vishwanath Venketaraman. 2020. "Understanding the Relationship between Glutathione, TGF-β, and Vitamin D in Combating Mycobacterium tuberculosis Infections" Journal of Clinical Medicine 9, no. 9: 2757. https://doi.org/10.3390/jcm9092757