The Role of Dendritic Cells in TB and HIV Infection

 , and
, and 
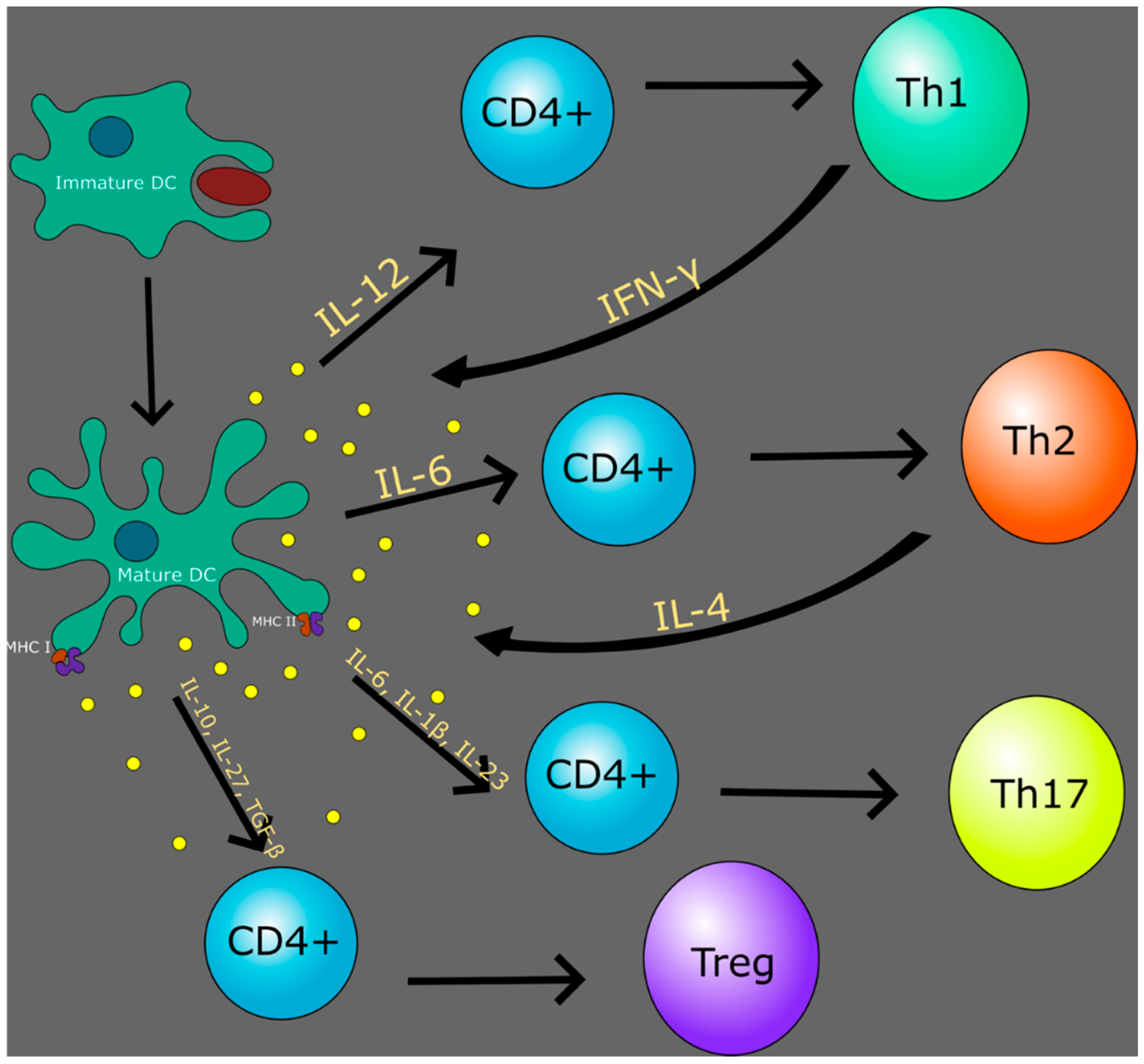{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Incidence and Epidemiology of Tuberculosis
2. Pathophysiology of Tuberculosis
3. Incidence and Epidemiology of HIV
4. Pathophysiology of HIV
5. Dendritic Cells
6. HIV and Dendritic Cells
7. Th1 vs. Th2 in Context of TB and HIV
8. Regulatory T Cells in Context of HIV and TB
9. Th17 Cells in Context of HIV and TB
10. Glutathione with Dendritic Cells to Improve Immune Response
11. The Role of GSH in Altering TB and HIV DC Functionality
12. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Harding, E. WHO global progress report on tuberculosis elimination. Lancet Respir. Med. 2020, 8, 19. [Google Scholar] [CrossRef]
- Ahmad, S. Pathogenesis, immunology, and diagnosis of latent mycobacterium tuberculosis infection. Clin. Dev. Immunol. 2011, 2011, 814943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaziou, P.; Sismanidis, C.; Floyd, K.; Raviglione, M. Global epidemiology of tuberculosis. Cold Spring Harb. Perspect. Med. 2014, 5, a017798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Crane, M.; Zhou, J.; Mina, M.; Post, J.J.; Cameron, B.A.; Lloyd, A.R.; Jaworowski, A.; French, M.A.; Lewin, S.R. HIV and co-infections. Immunol. Rev. 2013, 254, 114–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawn, S.D.; Wood, R.; De Cock, K.M.; Kranzer, K.; Lewis, J.J.; Churchyard, G.J. Antiretrovirals and isoniazid preventive therapy in the prevention of HIV-associated tuberculosis in settings with limited health-care resources. Lancet Infect. Dis. 2010, 10, 489–498. [Google Scholar] [CrossRef]
- Martinson, N.A.; Hoffmann, C.J.; Chaisson, R.E. Epidemiology of tuberculosis HIV recent advances in understanding responses. Proc. Am. Thorac. Soc. 2011, 8, 288–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, L. Revisiting the role of the granuloma in tuberculosis. Nat. Rev. Immunol. 2012, 12, 352–366. [Google Scholar] [CrossRef]
- Russell, D.G.; Cardona, P.J.; Kim, M.J.; Allain, S.; Altare, F. Foamy macrophages the progression of the human tuberculosis granuloma. Nat. Immunol. 2009, 10, 943–948. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.L.; Chan, J. Immunology of tuberculosis. Annu. Rev. Immunol. 2001, 19, 93–129. [Google Scholar] [CrossRef]
- Guirado, E.; Schlesinger, L.S. Modeling the mycobacterium tuberculosis granuloma-the critical battlefield in host immunity and disease. Front. Immunol. 2013, 4, 98. [Google Scholar] [CrossRef] [Green Version]
- Forrellad, M.A.; Klepp, L.I.; Gioffre, A.; Sabio y Garcia, J.; Morbidoni, H.R.; de la Paz Santangelo, M.; Cataldi, A.A.; Bigi, F. Virulence factors of the mycobacterium tuberculosis complex. Virulence 2013, 4, 3–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, C.; Pannecouque, C.; De Clercq, E.; Chen, F. Development of non-nucleoside reverse transcriptase inhibitors (NNRTIs): Our past twenty years. Acta Pharm. Sin. B 2020, 10, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Blood, German Advisory Committee. Human Immunodeficiency Virus (HIV). Transfus. Med. Hemother. 2016, 43, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Fettig, J.; Swaminathan, M.; Murrill, C.S.; Kaplan, J.E. Global epidemiology of HIV. Infect. Dis. Clin. N. Am. 2014, 28, 323–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Global HIV & AIDS Statistics—2020 Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 1 July 2020).
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef] [Green Version]
- HIV/AIDS. Available online: https://www.who.int/hiv/pub/guidelines/arv2013/intro/keyterms/en/ (accessed on 7 July 2020).
- Shaw, G.M.; Hunter, E. HIV transmission. Cold Spring Harb. Perspect. Med. 2012, 2, a006965. [Google Scholar] [CrossRef]
- Lackner, A.A.; Lederman, M.M.; Rodriguez, B. HIV pathogenesis: The host. Cold Spring Harb. Perspect. Med. 2012, 2, a007005. [Google Scholar] [CrossRef] [Green Version]
- Brenner, B.G.; Roger, M.; Routy, J.P.; Moisi, D.; Ntemgwa, M.; Matte, C.; Baril, J.G.; Thomas, R.; Rouleau, D.; Bruneau, J.; et al. High rates of forward transmission events after acute/early HIV-1 infection. J. Infect. Dis. 2007, 195, 951–959. [Google Scholar] [CrossRef]
- Miller, W.C.; Rosenberg, N.E.; Rutstein, S.E.; Powers, K.A. Role of acute and early HIV infection in the sexual transmission of HIV. Curr. Opin. HIV AIDS 2010, 5, 277–282. [Google Scholar] [CrossRef] [Green Version]
- Volz, E.M.; Ionides, E.; Romero-Severson, E.O.; Brandt, M.G.; Mokotoff, E.; Koopman, J.S. HIV-1 transmission during early infection in men who have sex with men: A phylodynamic analysis. PLoS Med. 2013, 10, e1001568. [Google Scholar] [CrossRef]
- Pierson, T.; McArthur, J.; Siliciano, R.F. Reservoirs for HIV-1: Mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu. Rev. Immunol. 2000, 18, 665–708. [Google Scholar] [CrossRef] [PubMed]
- Azevedo-Pereira, J.M.; Santos-Costa, Q. HIV interaction with human host: HIV-2 as a model of a less virulent infection. AIDS Rev. 2016, 18, 44–53. [Google Scholar] [PubMed]
- Campbell-Yesufu, O.T.; Gandhi, R.T. Update on human immunodeficiency virus (HIV)-2 infection. Clin. Infect. Dis. 2011, 52, 780–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugo-Villarino, G.; Cougoule, C.; Meunier, E.; Rombouts, Y.; Verollet, C.; Balboa, L. Editorial: The mononuclear phagocyte system in infectious disease. Front. Immunol. 2019, 10, 1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochando, J.; Kwan, W.H.; Ginhoux, F.; Hutchinson, J.A.; Hashimoto, D.; Collin, M. The mononuclear phagocyte system in organ transplantation. Am. J. Transplant. 2016, 16, 1053–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellman, I. Dendritic cells: Master regulators of the immune response. Cancer Immunol. Res. 2013, 1, 145–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Hammer, G.E.; Ma, A. Molecular control of steady-state dendritic cell maturation and immune homeostasis. Annu. Rev. Immunol. 2013, 31, 743–791. [Google Scholar] [CrossRef] [Green Version]
- Trombetta, E.S.; Mellman, I. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 2005, 23, 975–1028. [Google Scholar] [CrossRef]
- Morris, D.; Gonzalez, B.; Khurasany, M.; Kassissa, C.; Luong, J.; Kasko, S.; Pandya, S.; Chu, M.; Chi, P.T.; Bui, S.; et al. Characterization of dendritic cell and regulatory T cell functions against mycobacterium tuberculosis infection. Biomed. Res. Int. 2013, 2013, 402827. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Kim, T.H. Fundamental role of dendritic cells in inducing Th2 responses. Korean J. Intern. Med. 2018, 33, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terhune, J.; Berk, E.; Czerniecki, B.J. Dendritic cell-Induced Th1 and Th17 cell differentiation for cancer therapy. Vaccines 2013, 1, 527–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, R.; Hu, J. Role of dendritic cells in the induction of regulatory T cells. Cell Biosci. 2011, 1, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuka, M.; Egawa, G.; Kabashima, K. Uncovering the mysteries of langerhans cells, inflammatory dendritic epidermal cells; and monocyte-derived langerhans cell-like cells in the epidermis. Front. Immunol. 2018, 9, 1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deckers, J.; Hammad, H.; Hoste, E. Langerhans cells: Sensing the environment in health and disease. Front. Immunol. 2018, 9, 93. [Google Scholar] [CrossRef] [Green Version]
- Manches, O.; Frleta, D.; Bhardwaj, N. Dendritic cells in progression and pathology of HIV infection. Trends Immunol. 2014, 35, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Larsson, M. HIV-1 and the hijacking of dendritic cells: A tug of war. Springer Semin. Immunopathol. 2005, 26, 309–328. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, M.; Shen, Z.; Barr, F.D.; Boesch, A.W.; Ackerman, M.E.; Kappes, J.C.; Ochsenbauer, C.; Wira, C.R. Dendritic cells from the human female reproductive tract rapidly capture and respond to HIV. Mucosal Immunol. 2017, 10, 531–544. [Google Scholar] [CrossRef]
- Botting, R.A.; Rana, H.; Bertram, K.M.; Rhodes, J.W.; Baharlou, H.; Nasr, N.; Cunningham, A.L.; Harman, A.N. Langerhans cells and sexual transmission of HIV and HSV. Rev. Med. Virol. 2017, 27, e1923. [Google Scholar] [CrossRef]
- Matsuzawa, T.; Ogawa, Y.; Moriishi, K.; Shimada, S.; Kawamura, T. Immunological function of langerhans cells in HIV infection. J. Dermatol. Sci. 2017, 87, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena-Cruz, V.; Agosto, L.M.; Akiyama, H.; Olson, A.; Moreau, Y.; Larrieux, J.R.; Henderson, A.; Gummuluru, S.; Sagar, M. HIV-1 replicates and persists in vaginal epithelial dendritic cells. J. Clin. Investig. 2018, 128, 3439–3444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, R.I.; Sheridan, K.E.; Ceradini, D.; Choe, S.; Landau, N.R. Change in coreceptor use correlates with disease progression in HIV-1--infected individuals. J. Exp. Med. 1997, 185, 621–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, K.M.; Botting, R.A.; Baharlou, H.; Rhodes, J.W.; Rana, H.; Graham, J.D.; Patrick, E.; Fletcher, J.; Plasto, T.M.; Truong, N.R.; et al. Identification of HIV transmitting CD11c+ human epidermal dendritic cells. Nat. Commun. 2019, 10, 2759. [Google Scholar] [CrossRef]
- Lai, J.; Bernhard, O.K.; Turville, S.G.; Harman, A.N.; Wilkinson, J.; Cunningham, A.L. Oligomerization of the macrophage mannose receptor enhances gp120-mediated binding of HIV-1. J. Biol. Chem. 2009, 284, 11027–11038. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, Z.; Kawamura, T.; Shimada, S.; Piguet, V. The role of human dendritic cells in HIV-1 infection. J. Investig. Dermatol. 2015, 135, 1225–1233. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.; Bhardwaj, N. Dendritic cell dysregulation during HIV-1 infection. Immunol. Rev. 2013, 254, 170–189. [Google Scholar] [CrossRef] [Green Version]
- Su, B.; Biedma, M.E.; Lederle, A.; Peressin, M.; Lambotin, M.; Proust, A.; Decoville, T.; Schmidt, S.; Laumond, G.; Moog, C. Dendritic cell-lymphocyte cross talk downregulates host restriction factor SAMHD1 and stimulates HIV-1 replication in dendritic cells. J. Virol. 2014, 88, 5109–5121. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, E.; Cicala, C.; Van Ryk, D.; Goode, D.J.; Macleod, K.; Arthos, J.; Fauci, A.S. HIV-1 gp120 inhibits TLR9-mediated activation and IFN-{alpha} secretion in plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 2007, 104, 3396–3401. [Google Scholar] [CrossRef] [Green Version]
- Trifonova, R.T.; Bollman, B.; Barteneva, N.S.; Lieberman, J. Myeloid cells in intact human cervical explants capture HIV and can transmit it to CD4 T cells. Front. Immunol. 2018, 9, 2719. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; KewalRamani, V.N. Dendritic-cell interactions with HIV: Infection and viral dissemination. Nat. Rev. Immunol. 2006, 6, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Harman, A.N.; Wilkinson, J.; Bye, C.R.; Bosnjak, L.; Stern, J.L.; Nicholle, M.; Lai, J.; Cunningham, A.L. HIV induces maturation of monocyte-derived dendritic cells and Langerhans cells. J. Immunol. 2006, 177, 7103–7113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bess, J.W., Jr.; Gorelick, R.J.; Bosche, W.J.; Henderson, L.E.; Arthur, L.O. Microvesicles are a source of contaminating cellular proteins found in purified HIV-1 preparations. Virology 1997, 230, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavrois, M.; Neidleman, J.; Kreisberg, J.F.; Fenard, D.; Callebaut, C.; Greene, W.C. Human immunodeficiency virus fusion to dendritic cells declines as cells mature. J. Virol. 2006, 80, 1992–1999. [Google Scholar] [CrossRef] [Green Version]
- Mercier, S.K.; Donaghy, H.; Botting, R.A.; Turville, S.G.; Harman, A.N.; Nasr, N.; Ji, H.; Kusebauch, U.; Mendoza, L.; Shteynberg, D.; et al. The microvesicle component of HIV-1 inocula modulates dendritic cell infection and maturation and enhances adhesion to and activation of T lymphocytes. PLoS Pathog. 2013, 9, e1003700. [Google Scholar] [CrossRef]
- Fantuzzi, L.; Purificato, C.; Donato, K.; Belardelli, F.; Gessani, S. Human immunodeficiency virus type 1 gp120 induces abnormal maturation and functional alterations of dendritic cells: A novel mechanism for AIDS pathogenesis. J. Virol. 2004, 78, 9763–9772. [Google Scholar] [CrossRef] [Green Version]
- Hertoghs, N.; van der Aar, A.M.; Setiawan, L.C.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B. SAMHD1 degradation enhances active suppression of dendritic cell maturation by HIV-1. J. Immunol. 2015, 194, 4431–4437. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, D.G.; Bhardwaj, N. A division of labor: DC subsets and HIV receptor diversity. Nat. Immunol. 2002, 3, 891–893. [Google Scholar] [CrossRef]
- Rhodes, J.W.; Tong, O.; Harman, A.N.; Turville, S.G. Human dendritic cell subsets, ontogeny, and impact on HIV infection. Front. Immunol. 2019, 10, 1088. [Google Scholar] [CrossRef] [Green Version]
- Harman, A.N.; Bye, C.R.; Nasr, N.; Sandgren, K.J.; Kim, M.; Mercier, S.K.; Botting, R.A.; Lewin, S.R.; Cunningham, A.L.; Cameron, P.U. Identification of lineage relationships and novel markers of blood and skin human dendritic cells. J. Immunol. 2013, 190, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Bermejo-Jambrina, M.; Eder, J.; Helgers, L.C.; Hertoghs, N.; Nijmeijer, B.M.; Stunnenberg, M.; Geijtenbeek, T.B.H. C-type lectin receptors in antiviral immunity and viral escape. Front. Immunol. 2018, 9, 590. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.; Donkova-Petrini, V.; Caccavelli, L.; Balbo, M.; Carbonneil, C.; Levy, Y. Human immunodeficiency virus-driven expansion of CD4+CD25+ regulatory T cells, which suppress HIV-specific CD4 T-cell responses in HIV-infected patients. Blood 2004, 104, 3249–3256. [Google Scholar] [CrossRef] [PubMed]
- Abrahem, R.; Cao, R.; Robinson, B.; Munjal, S.; Cho, T.; To, K.; Ashley, D.; Hernandez, J.; Nguyen, T.; Teskey, G.; et al. Elucidating the efficacy of the bacille calmette-guerin vaccination in conjunction with first line antibiotics and liposomal glutathione. J. Clin. Med. 2019, 8, 1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, G.; Zhang, G.; Chen, X. Th1 cytokines; true functional signatures for protective immunity against TB? Cell. Mol. Immunol. 2018, 15, 206–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorman, S.E.; Holland, S.M. Interferon-gamma and interleukin-12 pathway defects and human disease. Cytokine Growth Factor Rev. 2000, 11, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Clerici, M.; Shearer, G.M. A TH1-->TH2 switch is a critical step in the etiology of HIV infection. Immunol. Today 1993, 14, 107–111. [Google Scholar] [CrossRef]
- Klein, S.A.; Dobmeyer, J.M.; Dobmeyer, T.S.; Pape, M.; Ottmann, O.G.; Helm, E.B.; Hoelzer, D.; Rossol, R. Demonstration of the Th1 to Th2 cytokine shift during the course of HIV-1 infection using cytoplasmic cytokine detection on single cell level by flow cytometry. AIDS 1997, 11, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Maggi, E.; Mazzetti, M.; Ravina, A.; Annunziato, F.; de Carli, M.; Piccinni, M.P.; Manetti, R.; Carbonari, M.; Pesce, A.M.; del Prete, G.; et al. Ability of HIV to promote a TH1 to TH0 shift and to replicate preferentially in TH2 and TH0 cells. Science 1994, 265, 244–248. [Google Scholar] [CrossRef]
- Yegorov, S.; Joag, V.; Galiwango, R.M.; Good, S.V.; Okech, B.; Kaul, R. Impact of endemic infections on HIV susceptibility in sub-saharan africa. Trop. Dis. Travel Med. Vaccines 2019, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Becker, Y. The changes in the T helper 1 (Th1) and T helper 2 (Th2) cytokine balance during HIV-1 infection are indicative of an allergic response to viral proteins that may be reversed by Th2 cytokine inhibitors and immune response modifiers—A review and hypothesis. Virus Genes 2004, 28, 5–18. [Google Scholar] [CrossRef]
- DiNardo, A.R.; Mandalakas, A.M.; Maphalala, G.; Mtetwa, G.; Mndzebele, T.; Ustero, P.; Hlatshwayo, M.; Mace, E.M.; Orange, J.S.; Makedonas, G. HIV Progression Perturbs the Balance of the Cell-Mediated and Anti-Inflammatory Adaptive and Innate Mycobacterial Immune Response. Mediat. Inflamm. 2016, 2016, 1478340. [Google Scholar] [CrossRef] [Green Version]
- Gulzar, N.; Copeland, K.F. CD8+ T-cells: Function and response to HIV infection. Curr. HIV Res. 2004, 2, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Okoye, A.A.; Picker, L.J. CD4+ T-cell depletion in HIV infection: Mechanisms of immunological failure. Immunol. Rev. 2013, 254, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaffen, S.L.; Liu, K.D. Overview of interleukin-2 function, production and clinical applications. Cytokine 2004, 28, 109–123. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, Y.; Zhang, S.; Luo, H.; Zhang, H. HIV-1 infection-induced suppression of the let-7i/IL-2 axis contributes to CD4+ T cell death. Sci. Rep. 2016, 6, 25341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geldmacher, C.; Koup, R.A. Pathogen-specific T cell depletion and reactivation of opportunistic pathogens in HIV infection. Trends Immunol. 2012, 33, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Getahun, H.; Gunneberg, C.; Granich, R.; Nunn, P. HIV infection-associated tuberculosis: The epidemiology and the response. Clin. Infect. Dis. 2010, 50 (Suppl. 3), S201–S207. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Ono, M.; Setoguchi, R.; Yagi, H.; Hori, S.; Fehervari, Z.; Shimizu, J.; Takahashi, T.; Nomura, T. Foxp3+CD25+CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 2006, 212, 8–27. [Google Scholar] [CrossRef]
- Angerami, M.T.; Suarez, G.V.; Vecchione, M.B.; Laufer, N.; Ameri, D.; Ben, G.; Perez, H.; Sued, O.; Salomon, H.; Quiroga, M.F. Expansion of CD25-negative forkhead box P3-positive T cells during HIV and mycobacterium tuberculosis Infection. Front. Immunol. 2017, 8, 528. [Google Scholar] [CrossRef] [Green Version]
- Fiorentino, D.F.; Zlotnik, A.; Vieira, P.; Mosmann, T.R.; Howard, M.; Moore, K.W.; O’Garra, A. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J. Immunol. 1991, 146, 3444–3451. [Google Scholar]
- Parkash, O.; Agrawal, S.; Madhan Kumar, M. T regulatory cells: Achilles’ heel of mycobacterium tuberculosis infection? Immunol. Res. 2015, 62, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Guyot-Revol, V.; Innes, J.A.; Hackforth, S.; Hinks, T.; Lalvani, A. Regulatory T cells are expanded in blood and disease sites in patients with tuberculosis. Am. J. Respir. Crit. Care Med. 2006, 173, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Sahmoudi, K.; Abbassi, H.; Bouklata, N.; El Alami, M.N.; Sadak, A.; Burant, C.; Henry Boom, W.; El Aouad, R.; Canaday, D.H.; Seghrouchni, F. Immune activation and regulatory T cells in mycobacterium tuberculosis infected lymph nodes. BMC Immunol. 2018, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Apoil, P.A.; Puissant, B.; Roubinet, F.; Abbal, M.; Massip, P.; Blancher, A. FOXP3 mRNA levels are decreased in peripheral blood CD4+ lymphocytes from HIV-positive patients. J. Acquir. Immune Defic. Syndr. 2005, 39, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Toor, J.S.; Singh, S.; Sharma, A.; Arora, S.K. Mycobacterium tuberculosis modulates the gene interactions to activate the HIV replication and faster disease progression in a co-infected host. PLoS ONE 2014, 9, e106815. [Google Scholar] [CrossRef] [Green Version]
- Lyadova, I.V.; Panteleev, A.V. Th1 and Th17 Cells in Tuberculosis: Protection; Pathology; and Biomarkers. Mediat. Inflamm. 2015, 2015, 854507. [Google Scholar] [CrossRef] [Green Version]
- Murray, L.W.; Satti, I.; Meyerowitz, J.; Jones, M.; Willberg, C.B.; Ussher, J.E.; Goedhals, D.; Hurst, J.; Phillips, R.E.; McShane, H.; et al. Human immunodeficiency virus infection impairs Th1 and Th17 mycobacterium tuberculosis-specific T-Cell responses. J. Infect. Dis. 2018, 217, 1782–1792. [Google Scholar] [CrossRef]
- Bixler, S.L.; Mattapallil, J.J. Loss and dysregulation of Th17 cells during HIV infection. Clin. Dev. Immunol. 2013, 2013, 852418. [Google Scholar] [CrossRef] [Green Version]
- Laan, M.; Cui, Z.H.; Hoshino, H.; Lotvall, J.; Sjostrand, M.; Gruenert, D.C.; Skoogh, B.E.; Linden, A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J. Immunol. 1999, 162, 2347–2352. [Google Scholar]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef]
- Oukka, M. Th17 cells in immunity and autoimmunity. Ann. Rheum. Dis. 2008, 67 (Suppl. 3), iii26–iii29. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, S.; Pizarro, T.T. The treg/Th17 axis: A dynamic balance regulated by the gut microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korb, V.C.; Phulukdaree, A.; Lalloo, U.G.; Chuturgoon, A.A.; Moodley, D. TB/HIV pleurisy reduces Th17 lymphocyte proportion independent of the cytokine microenvironment. Tuberculosis (Edinb.) 2016, 99, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieh, D.J.; Matias, E.; Xu, H.; Fought, A.J.; Blanchard, J.L.; Marx, P.A.; Veazey, R.S.; Hope, T.J. Th17 Cells Are Preferentially Infected Very Early after Vaginal Transmission of SIV in Macaques. Cell Host Microbe 2016, 19, 529–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planas, D.; Zhang, Y.; Monteiro, P.; Goulet, J.P.; Gosselin, A.; Grandvaux, N.; Hope, T.J.; Fassati, A.; Routy, J.P.; Ancuta, P. HIV-1 selectively targets gut-homing CCR6+CD4+ T cells via mTOR-dependent mechanisms. JCI Insight 2017, 2, e93230. [Google Scholar] [CrossRef]
- Fernandes, J.R.; Berthoud, T.K.; Kumar, A.; Angel, J.B. IL-23 signaling in Th17 cells is inhibited by HIV infection and is not restored by HAART: Implications for persistent immune activation. PLoS ONE 2017, 12, e0186823. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Oestreicher, J.; Morgan, B. Glutathione: Subcellular distribution and membrane transport. Biochem. Cell Biol. 2019, 97, 270–289. [Google Scholar] [CrossRef] [Green Version]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc− cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Kamide, Y.; Utsugi, M.; Dobashi, K.; Ono, A.; Ishizuka, T.; Hisada, T.; Koga, Y.; Uno, K.; Hamuro, J.; Mori, M. Intracellular glutathione redox status in human dendritic cells regulates IL-27 production and T-cell polarization. Allergy 2011, 66, 1183–1192. [Google Scholar] [CrossRef]
- Yan, Z.; Banerjee, R. Redox remodeling as an immunoregulatory strategy. Biochemistry 2010, 49, 1059–1066. [Google Scholar] [PubMed] [Green Version]
- Alam, K.; Ghousunnissa, S.; Nair, S.; Valluri, V.L.; Mukhopadhyay, S. Glutathione-redox balance regulates c-rel-driven IL-12 production in macrophages possible implications in antituberculosis immunotherapy. J. Immunol. 2010, 184, 2918–2929. [Google Scholar] [CrossRef] [PubMed]
- Short, S.; Merkel, B.J.; Caffrey, R.; McCoy, K.L. Defective antigen processing correlates with a low level of intracellular glutathione. Eur. J. Immunol. 1996, 26, 3015–3020. [Google Scholar] [CrossRef] [PubMed]
- Mizuochi, T.; Yee, S.T.; Kasai, M.; Kakiuchi, T.; Muno, D.; Kominami, E. Both cathepsin B and cathepsin D are necessary for processing of ovalbumin as well as for degradation of class II MHC invariant chain. Immunol. Lett. 1994, 43, 189–193. [Google Scholar] [CrossRef]
- Vignali, D.A.; Kuchroo, V.K. IL-12 family cytokines: Immunological playmakers. Nat. Immunol. 2012, 13, 722–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, A.; Kaushal, P.; Agrawal, S.; Gollapudi, S.; Gupta, S. Thimerosal induces TH2 responses via influencing cytokine secretion by human dendritic cells. J. Leukoc Biol. 2007, 81, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Tailleux, L.; Schwartz, O.; Herrmann, J.L.; Pivert, E.; Jackson, M.; Amara, A.; Legres, L.; Dreher, D.; Nicod, L.P.; Gluckman, J.C.; et al. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J. Exp. Med. 2003, 197, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Angelini, G.; Gardella, S.; Ardy, M.; Ciriolo, M.R.; Filomeni, G.; Di Trapani, G.; Clarke, F.; Sitia, R.; Rubartelli, A. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc. Natl. Acad. Sci. USA 2002, 99, 1491–1496. [Google Scholar] [CrossRef] [Green Version]
- Benhar, M.; Shytaj, I.L.; Stamler, J.S.; Savarino, A. Dual targeting of the thioredoxin and glutathione systems in cancer and HIV. J. Clin. Investig. 2016, 126, 1630–1639. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell. Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.; Ly, J.; Chi, P.T.; Daliva, J.; Nguyen, T.; Soofer, C.; Chen, Y.C.; Lagman, M.; Venketaraman, V. Glutathione synthesis is compromised in erythrocytes from individuals with HIV. Front. Pharmacol. 2014, 5, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, S.C.; Zaretsky, M.D.; Dubs, J.G.; Roederer, M.; Anderson, M.; Green, A.; Mitra, D.; Watanabe, N.; Nakamura, H.; Tjioe, I.; et al. N-acetylcysteine replenishes glutathione in HIV infection. Eur. J. Clin. Investig. 2000, 30, 915–929. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abrahem, R.; Chiang, E.; Haquang, J.; Nham, A.; Ting, Y.-S.; Venketaraman, V. The Role of Dendritic Cells in TB and HIV Infection. J. Clin. Med. 2020, 9, 2661. https://doi.org/10.3390/jcm9082661
Abrahem R, Chiang E, Haquang J, Nham A, Ting Y-S, Venketaraman V. The Role of Dendritic Cells in TB and HIV Infection. Journal of Clinical Medicine. 2020; 9(8):2661. https://doi.org/10.3390/jcm9082661
Chicago/Turabian StyleAbrahem, Rachel, Emerald Chiang, Joseph Haquang, Amy Nham, Yu-Sam Ting, and Vishwanath Venketaraman. 2020. "The Role of Dendritic Cells in TB and HIV Infection" Journal of Clinical Medicine 9, no. 8: 2661. https://doi.org/10.3390/jcm9082661