The Future of Lipid-Lowering Therapy


Abstract
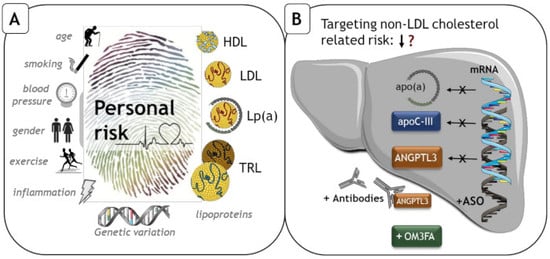
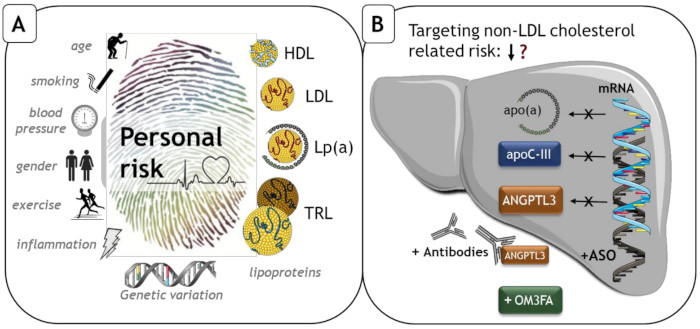
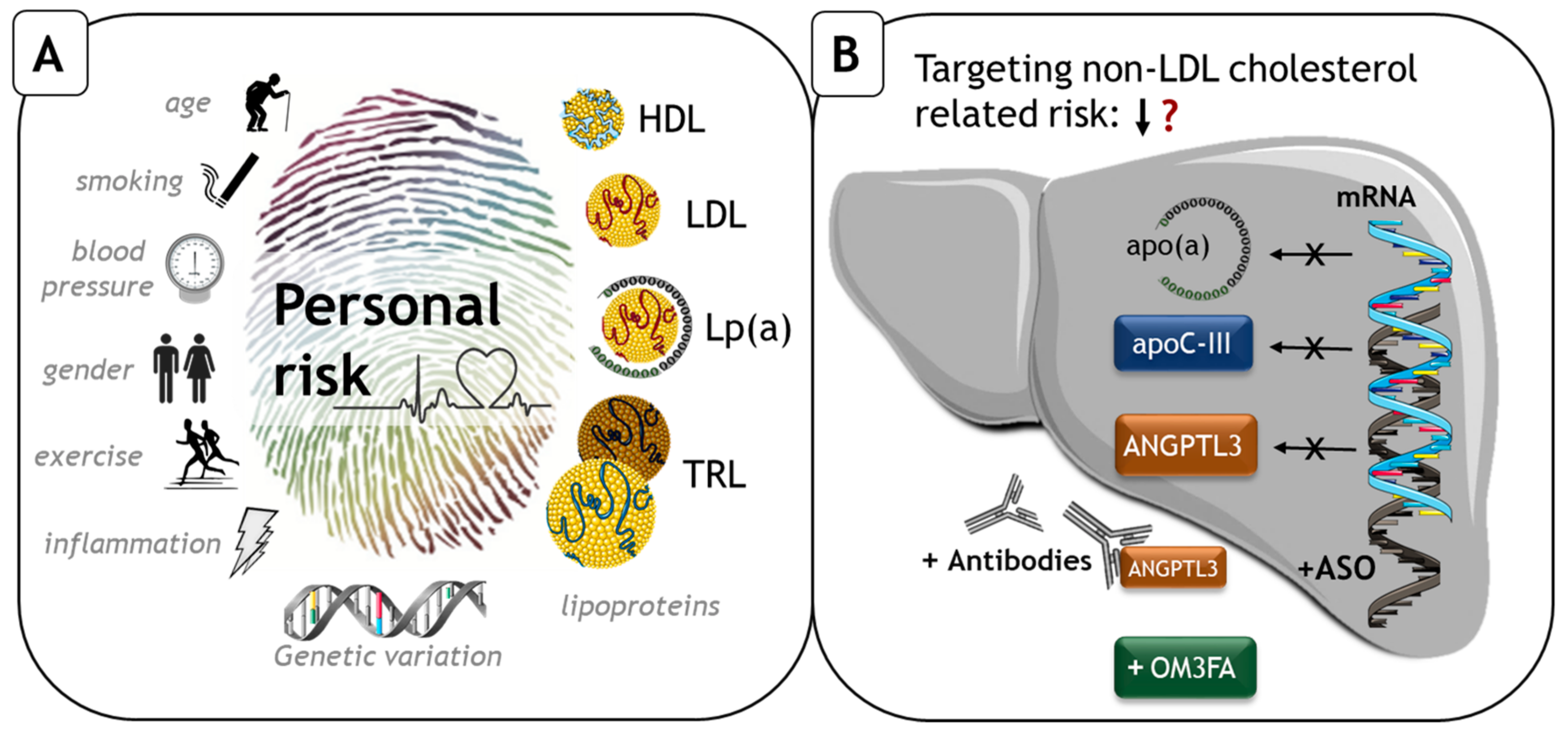
:
1. Introduction
2. LPA, Apolipoprotein (a), Lp(a)
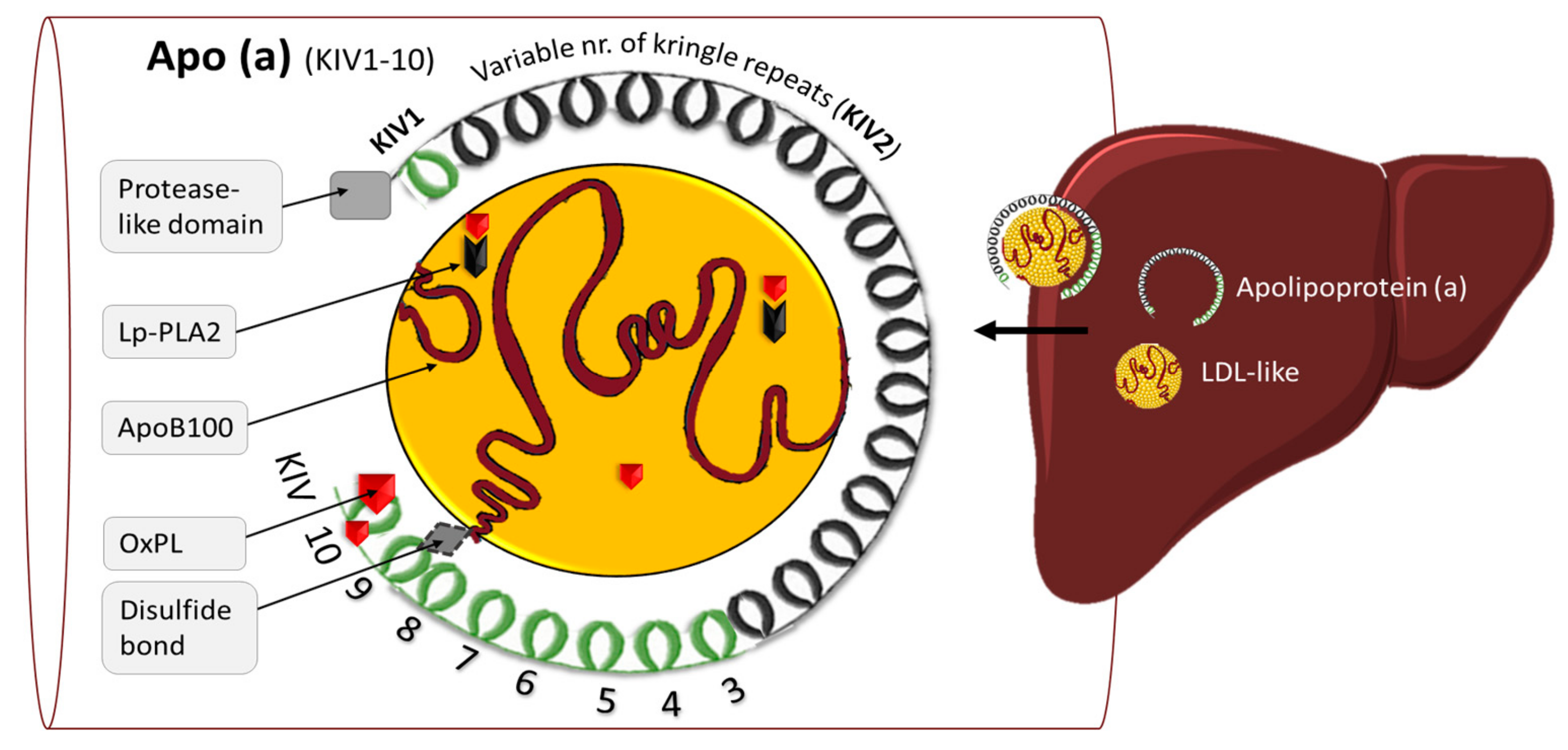
2.1. Structure and Function
2.2. Observational
2.3. Drug Development
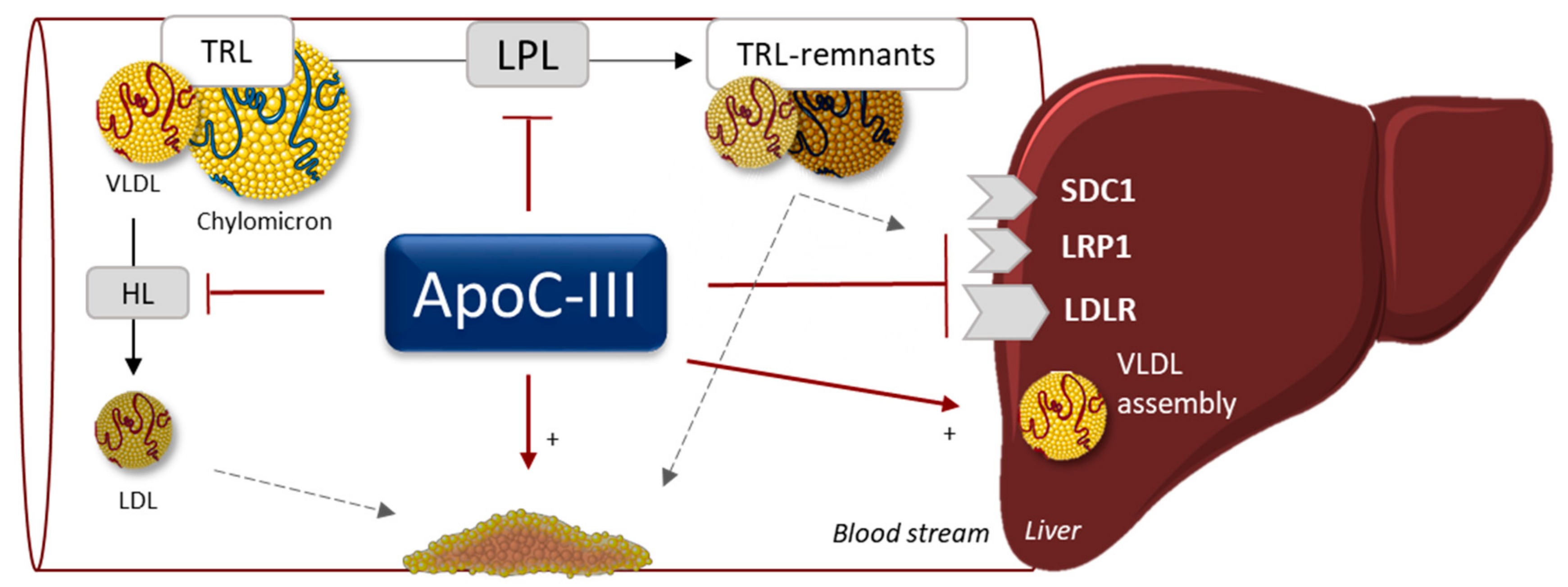
3. APOC3, apoC-III
3.1. Function
3.2. Observational
3.3. Drug Development
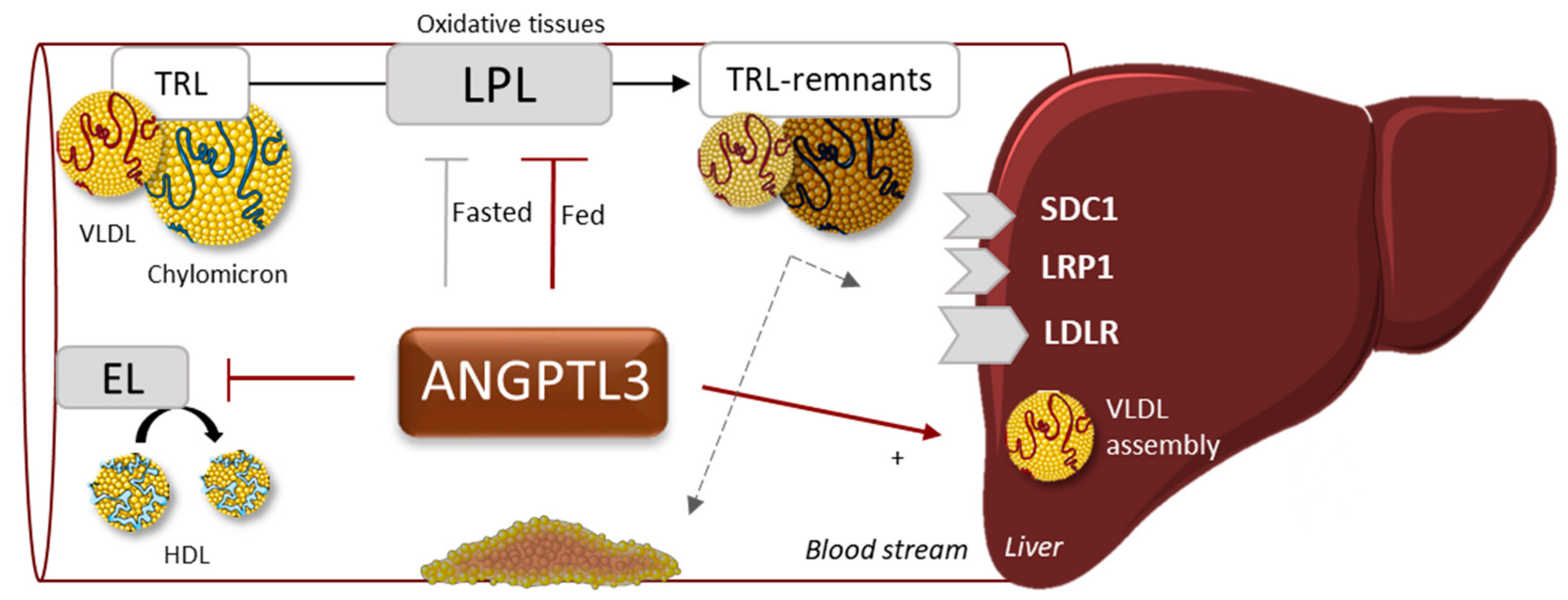
4. ANGPTL3, Angiopoietin-Like Protein 3
4.1. Function
4.2. Observational
4.3. Drug Development
5. Omega 3 Fatty Acids
5.1. Type and Function
5.2. Observational
5.3. Drug Development
6. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Sidney, S.; Quesenberry, C.P.; Jaffe, M.G.; Sorel, M.; Nguyen-Huynh, M.N.; Kushi, L.H.; Go, A.S.; Rana, J.S. Recent Trends in Cardiovascular Mortality in the United States and Public Health Goals. JAMA Cardiol. 2016, 1, 594. [Google Scholar] [CrossRef]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions. JAMA 2016, 316, 1289. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.D. Inadequate control of atherosclerotic cardiovascular disease risk factors in Europe: EUROASPIRE repeats itself. Eur. J. Prev. Cardiol. 2019, 26, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. Ann. Clin. Biochem. Int. J. Lab. Med. 2017, 54, 511. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Kamstrup, P.R. Genetically Elevated Lipoprotein(a) and Increased Risk of Myocardial Infarction. JAMA 2009, 301, 2331. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.C.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S.; et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 2009, 361, 2518–2528. [Google Scholar] [CrossRef]
- Do, R.; Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Gao, C.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat. Genet. 2013, 45, 1345–1352. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Toth, P.P.; Granowitz, C.; Hull, M.; Liassou, D.; Anderson, A.; Philip, S. High Triglycerides Are Associated With Increased Cardiovascular Events, Medical Costs, and Resource Use: A Real-World Administrative Claims Analysis of Statin-Treated Patients With High Residual Cardiovascular Risk. J. Am. Heart Assoc. 2018, 7, e008740. [Google Scholar] [CrossRef] [PubMed]
- Endo, J.; Arita, M. Cardioprotective mechanism of omega-3 polyunsaturated fatty acids. J. Cardiol. 2016, 67, 22–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Ionis Pharmaceuticals and/or Akcea Therapeutics Akcea and Ionis Report Positive Data from Phase 2 Study of AKCEA-APO(a)-LRx. Available online: https://ir.ionispharma.com/news-releases/news-release-details/phase-2-results-akcea-apoa-lrx-presented-late-breaking-clinical (accessed on 22 April 2019).
- Gouni-Berthold, I.; Alexander, V.; Digenio, A.; DuFour, R.; Steinhagen-Thiessen, E.; Martin, S.; Moriarty, P.; Hughes, S.; Witztum, J.; Gaudet, D.; et al. Apolipoprotein C-III Inhibition With Volanesorsen in Patients With Hypertriglyceridemia (COMPASS): A Randomized, Double-Blind, Placebo-Controlled Trial. J. Clin. Lipidol. 2017, 11, 794–795. [Google Scholar] [CrossRef]
- Alexander, V.J.; Digenio, A.; Xia, S.; Hurh, E.; Hughes, S.; Geary, R.S.; Witztum, J.L.; Tsimikas, S. Inhibition of Apolipoprotein C-III with GalNAc-Conjugated Antisense Drug Potently Lowers Fasting Serum Apolipoprotein C-III and Triglyceride Levels in Healthy Volunteers with Elevated Triglycerides. J. Am. Coll. Cardiol. 2018, 71, A1724. [Google Scholar] [CrossRef]
- Ahmad, Z.; Banerjee, P.; Hamon, S.; Chan, K.-C.; Bouzelmat, A.; Sasiela, W.J.; Pordy, R.; Mellis, S.; Dansky, H.; Gipe, D.A.; et al. Inhibition of Angiopoietin-Like Protein 3 With a Monoclonal Antibody Reduces Triglycerides in Hypertriglyceridemia. Circulation 2019. [Google Scholar] [CrossRef]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef]
- Backes, J.; Anzalone, D.; Hilleman, D.; Catini, J. The clinical relevance of omega-3 fatty acids in the management of hypertriglyceridemia. Lipids Health Dis. 2016, 15, 118. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef]
- Trieu, V.N.; McConathy, W.J. A two-step model for lipoprotein(a) formation. J. Biol. Chem. 1995, 270, 15471–15474. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Albers, J.J.; Gabel, B.; Koschinsky, M.L.; Gaur, V.P. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin. Chem. 1995, 41, 246–255. [Google Scholar] [PubMed]
- Tsimikas, S. A Test in Context: Lipoprotein(a). J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef] [PubMed]
- Boerwinkle, E.; Leffert, C.C.; Lin, J.; Lackner, C.; Chiesa, G.; Hobbs, H.H. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J. Clin. Investig. 1992, 90, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Langsted, A. Lipoprotein (a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J. Lipid Res. 2016, 57, 1953–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibundgut, G.; Scipione, C.; Yin, H.; Schneider, M.; Boffa, M.B.; Green, S.; Yang, X.; Dennis, E.; Witztum, J.L.; Koschinsky, M.L.; et al. Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a). J. Lipid Res. 2013, 54, 2815–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmark, C.; Dewan, A.; Orsoni, A.; Merki, E.; Miller, E.R.; Shin, M.-J.; Binder, C.J.; Hörkkö, S.; Krauss, R.M.; Chapman, M.J.; et al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J. Lipid Res. 2008, 49, 2230–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Steeg, W.A.; Holme, I.; Boekholdt, S.M.; Larsen, M.L.; Lindahl, C.; Stroes, E.S.G.; Tikkanen, M.J.; Wareham, N.J.; Faergeman, O.; Olsson, A.G.; et al. High-Density Lipoprotein Cholesterol, High-Density Lipoprotein Particle Size, and Apolipoprotein A-I: Significance for Cardiovascular Risk. J. Am. Coll. Cardiol. 2008, 51, 634–642. [Google Scholar] [CrossRef]
- Boffelli, D.; Cheng, J.F.; Rubin, E.M. Convergent evolution in primates and an insectivore. Genomics 2004. [Google Scholar] [CrossRef]
- Yeang, C.; Cotter, B.; Tsimikas, S. Experimental Animal Models Evaluating the Causal Role of Lipoprotein(a) in Atherosclerosis and Aortic Stenosis. Cardiovasc. Drugs Ther. 2016, 30, 75–85. [Google Scholar] [CrossRef]
- Tsimikas, S.; Fazio, S.; Ferdinand, K.C.; Ginsberg, H.N.; Koschinsky, M.L.; Marcovina, S.M.; Moriarty, P.M.; Rader, D.J.; Remaley, A.T.; Reyes-Soffer, G.; et al. NHLBI Working Group Recommendations to Reduce Lipoprotein(a)-Mediated Risk of Cardiovascular Disease and Aortic Stenosis. J. Am. Coll. Cardiol. 2018, 71, 177–192. [Google Scholar] [CrossRef]
- Emerging Risk Factors Collaboration; Erqou, S.; Kaptoge, S.; Perry, P.L.; Di Angelantonio, E.; Thompson, A.; White, I.R.; Marcovina, S.M.; Collins, R.; Thompson, S.G.; et al. Lipoprotein(a) Concentration and the Risk of Coronary Heart Disease, Stroke, and Nonvascular Mortality. JAMA 2009, 302, 412. [Google Scholar] [PubMed]
- Langsted, A.; Kamstrup, P.R.; Nordestgaard, B.G. High lipoprotein(a) and high risk of mortality. Eur. Heart J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Benn, M.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: The Copenhagen City Heart Study. Circulation 2008, 117, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Thanassoulis, G.; Campbell, C.Y.; Owens, D.S.; Smith, J.G.; Smith, A.V.; Peloso, G.M.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic Associations with Valvular Calcification and Aortic Stenosis. N. Engl. J. Med. 2013, 368, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsenault, B.J.; Boekholdt, S.M.; Dubé, M.-P.; Rhéaume, É.; Wareham, N.J.; Khaw, K.-T.; Sandhu, M.S.; Tardif, J.-C. Lipoprotein(a) Levels, Genotype, and Incident Aortic Valve Stenosis. Circ. Cardiovasc. Genet. 2014, 7, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Li, X.; Feng, Q.; Kubo, M.; Kullo, I.J.; Peissig, P.L.; Karlson, E.W.; Jarvik, G.P.; Lee, M.T.M.; Shang, N.; et al. LPA Variants Are Associated With Residual Cardiovascular Risk in Patients Receiving Statins. Circulation 2018, 138, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Lamina, C.; Kronenberg, F. Estimation of the Required Lipoprotein(a)-Lowering Therapeutic Effect Size for Reduction in Coronary Heart Disease Outcomes. JAMA Cardiol. 2019. [Google Scholar] [CrossRef]
- Kinpara, K.; Okada, H.; Yoneyama, A.; Okubo, M.; Murase, T. Lipoprotein(a)-cholesterol: A significant component of serum cholesterol. Clin. Chim. Acta 2011, 412, 1783–1787. [Google Scholar] [CrossRef]
- Yeang, C.; Witztum, J.L.; Tsimikas, S. ‘LDL-C’ = LDL-C + Lp(a)-C. Curr. Opin. Lipidol. 2015, 26, 169–178. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Borén, J.; Andreotti, F.; Watts, G.F.; Ginsberg, H.; Amarenco, P.; Catapano, A.; Descamps, O.S.; et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur. Heart J. 2010, 31, 2844–2853. [Google Scholar] [CrossRef]
- Verbeek, R.; Boekholdt, S.M.; Stoekenbroek, R.M.; Hovingh, G.K.; Witztum, J.L.; Wareham, N.J.; Sandhu, M.S.; Khaw, K.-T.; Tsimikas, S. Population and assay thresholds for the predictive value of lipoprotein (a) for coronary artery disease: the EPIC-Norfolk Prospective Population Study. J. Lipid Res. 2016, 57, 697–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, W.; Cao, J.; Steffen, B.T.; Post, W.S.; Stein, J.H.; Tattersall, M.C.; Kaufman, J.D.; McConnell, J.P.; Hoefner, D.M.; Warnick, R.; et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: The multi-ethnic study of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Paré, G.; Çaku, A.; McQueen, M.; Anand, S.S.; Enas, E.; Clarke, R.; Boffa, M.B.; Koschinsky, M.; Wang, X.; Yusuf, S. Lipoprotein(a) Levels and the Risk of Myocardial Infarction Among 7 Ethnic Groups. Circulation 2019, 139, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Varvel, S.; McConnell, J.P.; Tsimikas, S. Prevalence of Elevated Lp(a) Mass Levels and Patient Thresholds in 532 359 Patients in the United States. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2239–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, R.; Andres, E.; Mata, N.; Fuentes-Jiménez, F.; Badimón, L.; López-Miranda, J.; Padró, T.; Muñiz, O.; Díaz-Díaz, J.L.; Mauri, M.; et al. Lipoprotein(a) levels in familial hypercholesterolemia: An important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J. Am. Coll. Cardiol. 2014, 63, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2019, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stiekema, L.C.A.; Stroes, E.S.G.; Verweij, S.L.; Kassahun, H.; Chen, L.; Wasserman, S.M.; Sabatine, M.S.; Mani, V.; Fayad, Z.A. Persistent arterial wall inflammation in patients with elevated lipoprotein(a) despite strong low-density lipoprotein cholesterol reduction by proprotein convertase subtilisin/kexin type 9 antibody treatment. Eur. Heart J. 2018, 0, 1–8. [Google Scholar] [CrossRef]
- Ito, Y.; Azrolan, N.; O’Connell, A.; Walsh, A.; Breslow, J.L. Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science 1990, 249, 790–793. [Google Scholar] [CrossRef]
- Maeda, N.; Li, H.; Lee, D.; Oliver, P.; Quarfordt, S.H.; Osada, J. Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. J. Biol. Chem. 1994, 269, 23610–23616. [Google Scholar]
- Ginsberg, H.N.; Le, N.A.; Goldberg, I.J.; Gibson, J.C.; Rubinstein, A.; Wang-Iverson, P.; Norum, R.; Brown, W.V. Apolipoprotein B metabolism in subjects with deficiency of apolipoproteins CIII and AI. Evidence that apolipoprotein CIII inhibits catabolism of triglyceride-rich lipoproteins by lipoprotein lipase in vivo. J. Clin. Investig. 1986, 78, 1287–1295. [Google Scholar] [CrossRef]
- Sundaram, M.; Zhong, S.; Khalil, M.B.; Links, P.H.; Zhao, Y.; Iqbal, J.; Hussain, M.M.; Parks, R.J.; Wang, Y.; Yao, Z. Expression of apolipoprotein C-III in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. J. Lipid Res. 2010, 51, 150–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, C.E.; Osborne, J.C.; Schaefer, E.J.; Brewer, H.B. Activation of the Enzymic Activity of Hepatic Lipase by Apolipoprotein A-II. Characterization of a Major Component of High Density Lipoprotein as the Activating Plasma Component in vitro. Eur. J. Biochem. 1983, 131, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.; Curtis, K.R.; Amir Alipour, M.; LeBlond, N.D.; Margison, K.D.; Yaworski, R.A.; Parks, R.J.; McIntyre, A.D.; Hegele, R.A.; Fullerton, M.D.; et al. The apolipoprotein C-III (Gln38Lys) variant associated with human hypertriglyceridemia is a gain-of-function mutation. J. Lipid Res. 2017, 58, 2188–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordts, P.L.S.M.; Nock, R.; Son, N.-H.; Ramms, B.; Lew, I.; Gonzales, J.C.; Thacker, B.E.; Basu, D.; Lee, R.G.; Mullick, A.E.; et al. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J. Clin. Investig. 2016, 126, 2855–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khetarpal, S.A.; Zeng, X.; Millar, J.S.; Vitali, C.; Somasundara, A.V.H.; Zanoni, P.; Landro, J.A.; Barucci, N.; Zavadoski, W.J.; Sun, Z.; et al. A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat. Med. 2017, 23, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Ramms, B.; Patel, S.; Nora, C.; Pessentheiner, A.R.; Chang, M.W.; Green, C.R.; Golden, G.J.; Secrest, P.; Krauss, R.M.; Metallo, C.M.; et al. ApoC-III ASO Promotes Tissue LPL Activity in Absence of ApoE-Mediated TRL Clearance. J. Lipid Res. 2019, jlr.M093740. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M. The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia. Curr. Opin. Lipidol. 2015, 26, 56–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehayek, E.; Eisenberg, S. Mechanisms of inhibition by apolipoprotein C of apolipoprotein E-dependent cellular metabolism of human triglyceride-rich lipoproteins through the low density lipoprotein receptor pathway. J. Biol. Chem. 1991, 266, 18259–18267. [Google Scholar]
- Kawakami, A.; Aikawa, M.; Alcaide, P.; Luscinskas, F.W.; Libby, P.; Sacks, F.M. Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation 2006, 114, 681–687. [Google Scholar] [CrossRef]
- Åvall, K.; Ali, Y.; Leibiger, I.B.; Leibiger, B.; Moede, T.; Paschen, M.; Dicker, A.; Daré, E.; Köhler, M.; Ilegems, E.; et al. Apolipoprotein CIII links islet insulin resistance to β-cell failure in diabetes. Proc. Natl. Acad. Sci. USA 2015, 112, E2611–E2619. [Google Scholar] [CrossRef]
- Taskinen, M.; Packard, C.J.; Borén, J. Emerging Evidence that ApoC-III Inhibitors Provide Novel Options to Reduce the Residual CVD. Curr. Atheroscler. Rep. 2019, 21, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clive, R.; Pullinger, M.J.; Malloy, A.K.; Shahidi, M.G.; Duchateau, P.; Villagomez, J.; Allaart, J.J.P.; Kane, J.P. A novel apolipoprotein C-Ill variant, apoC-III(Gln38 -+ Lys), associated with moderate hypertriglyceridemia in a large kindred of Mexican origin. J. Lipid Res. 1997, 38, 1833–1840. [Google Scholar]
- TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute. Loss-of-Function Mutations in APOC3, Triglycerides, and Coronary Disease. N. Engl. J. Med. 2014, 371, 22–31. [Google Scholar] [PubMed]
- Reyes-Soffer, G.; Sztalryd, C.; Horenstein, R.B.; Holleran, S.; Matveyenko, A.; Thomas, T.; Nandakumar, R.; Ngai, C.; Karmally, W.; Ginsberg, H.N.; et al. Effects of APOC3 Heterozygous Deficiency on Plasma Lipid and Lipoprotein Metabolism. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Saleheen, D.; Natarajan, P.; Armean, I.M.; Zhao, W.; Rasheed, A.; Khetarpal, S.A.; Won, H.H.; Karczewski, K.J.; O’Donnell-Luria, A.H.; Samocha, K.E.; et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature 2017, 544, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olkkonen, V.M.; Sinisalo, J.; Jauhiainen, M. New medications targeting triglyceride-rich lipoproteins: Can inhibition of ANGPTL3 or apoC-III reduce the residual cardiovascular risk? Atherosclerosis 2018, 272, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudet, D.; Alexander, V.J.; Baker, B.F.; Brisson, D.; Tremblay, K.; Singleton, W.; Geary, R.S.; Hughes, S.G.; Viney, N.J.; Graham, M.J. Antisense Inhibition of Apolipoprotein C-III in Patients with Hypertriglyceridemia. N. Engl. J. Med. 2015, 373, 438–447. [Google Scholar] [CrossRef]
- Digenio, A.; Dunbar, R.L.; Alexander, V.J.; Hompesch, M.; Morrow, L.; Lee, R.G.; Graham, M.J.; Hughes, S.G.; Yu, R.; Singleton, W.; et al. Antisense-Mediated Lowering of Plasma Apolipoprotein C-III by Volanesorsen Improves Dyslipidemia and Insulin Sensitivity in Type 2 Diabetes. Diabetes Care 2016, 39, 1408–1415. [Google Scholar] [CrossRef] [Green Version]
- Gaudet, D.; Digenio, A.; Alexander, V.J.; Arca, M.; Jones, A.F.; Stroes, E.; Bergeron, J.; Civeira, F.; Hemphill, L.; Blom, D.J.; et al. The APPROACH Study: A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Study of Volanesorsen Administered Subcutaneously to Patients with Familial Chylomicronemia Syndrome (FCS). J. Clin. Lipidol. 2017, 11, 814–815. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use (CHMP). Assessment Report Volanosorsen/waylivra; European Medicines Agency: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Akcea Therapeutics Study of ISIS 678354 (AKCEA-APOCIII-LRx) in Patients With Hypertriglyceridemia and Established Cardiovascular Disease (CVD). Available online: https://www.clinicaltrials.gov/ct2/show/NCT03385239?term=AKCEA-APOCIII-LRx&rank=1 (accessed on 21 February 2019).
- Koishi, R.; Ando, Y.; Ono, M.; Shimamura, M.; Yasumo, H.; Fujiwara, T.; Horikoshi, H.; Furukawa, H. Angptl3 regulates lipid metabolism in mice. Nat. Genet. 2002, 30, 151–157. [Google Scholar] [CrossRef]
- Shimamura, M.; Matsuda, M.; Yasumo, H.; Okazaki, M.; Fujimoto, K.; Kono, K.; Shimizugawa, T.; Ando, Y.; Koishi, R.; Kohama, T.; et al. Angiopoietin-Like Protein3 Regulates Plasma HDL Cholesterol Through Suppression of Endothelial Lipase. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 366–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.-C.; Desai, U.; Gololobov, G.; Hong, S.; Feng, X.; Yu, X.-C.; Gay, J.; Wilganowski, N.; Gao, C.; Du, L.-L.; et al. Identification of a New Functional Domain in Angiopoietin-like 3 (ANGPTL3) and Angiopoietin-like 4 (ANGPTL4) Involved in Binding and Inhibition of Lipoprotein Lipase (LPL). J. Biol. Chem. 2009, 284, 13735–13745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gusarova, V.; Banfi, S.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Inactivation of ANGPTL3 reduces hepatic VLDL-triglyceride secretion. J. Lipid Res. 2015, 56, 1296–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisciotta, L.; Favari, E.; Magnolo, L.; Simonelli, S.; Adorni, M.P.; Sallo, R.; Fancello, T.; Zavaroni, I.; Ardigò, D.; Bernini, F.; et al. Characterization of Three Kindreds With Familial Combined Hypolipidemia Caused by Loss-of-Function Mutations of ANGPTL3. Circ. Cardiovasc. Genet. 2012, 5, 42–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musunuru, K.; Pirruccello, J.P.; Do, R.; Peloso, G.M.; Guiducci, C.; Sougnez, C.; Garimella, K.V.; Fisher, S.; Abreu, J.; Barry, A.J.; et al. Exome Sequencing, ANGPTL3 Mutations, and Familial Combined Hypolipidemia. N. Engl. J. Med. 2010, 363, 2220–2227. [Google Scholar] [CrossRef]
- Minicocci, I.; Montali, A.; Robciuc, M.R.; Quagliarini, F.; Censi, V.; Labbadia, G.; Gabiati, C.; Pigna, G.; Sepe, M.L.; Pannozzo, F.; et al. Mutations in the ANGPTL3 Gene and Familial Combined Hypolipidemia: A Clinical and Biochemical Characterization. J. Clin. Endocrinol. Metab. 2012, 97, E1266–E1275. [Google Scholar] [CrossRef] [PubMed]
- Robciuc, M.R.; Maranghi, M.; Lahikainen, A.; Rader, D.; Bensadoun, A.; Öörni, K.; Metso, J.; Minicocci, I.; Ciociola, E.; Ceci, F.; et al. Angptl3 Deficiency Is Associated With Increased Insulin Sensitivity, Lipoprotein Lipase Activity, and Decreased Serum Free Fatty Acids. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1706–1713. [Google Scholar] [CrossRef] [Green Version]
- Stitziel, N.O.; Khera, A.V.; Wang, X.; Bierhals, A.J.; Vourakis, A.C.; Sperry, A.E.; Natarajan, P.; Klarin, D.; Emdin, C.A.; Zekavat, S.M.; et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69, 2054–2063. [Google Scholar] [CrossRef]
- Gaudet, D.; Gipe, D.A.; Pordy, R.; Ahmad, Z.; Cuchel, M.; Shah, P.K.; Chyu, K.-Y.; Sasiela, W.J.; Chan, K.-C.; Brisson, D.; et al. ANGPTL3 Inhibition in Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2017, 377, 296–297. [Google Scholar] [CrossRef]
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.-J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; McEvoy, B.W.; et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232. [Google Scholar] [CrossRef]
- Davidson, M.H.; Johnson, J.; Rooney, M.W.; Kyle, M.L.; Kling, D.F. A novel omega-3 free fatty acid formulation has dramatically improved bioavailability during a low-fat diet compared with omega-3-acid ethyl esters: The ECLIPSE (Epanova® compared to Lovaza® in a pharmacokinetic single-dose evaluation) study. J. Clin. Lipidol. 2012, 6, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Dyerberg, J.; Madsen, P.; Møller, J.M.; Aardestrup, I.; Schmidt, E.B. Bioavailability of marine n-3 fatty acid formulations. Prostaglandins Leukot. Essent. Fat. Acids 2010, 83, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.C.; Bobotas, G.; Dicklin, M.R.; Huebner, M.; Keane, W.F. Effects of MAT9001 containing eicosapentaenoic acid and docosapentaenoic acid, compared to eicosapentaenoic acid ethyl esters, on triglycerides, lipoprotein cholesterol, and related variables. J. Clin. Lipidol. 2017, 11, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.Y.; Jacobson, T.A. Effects of Eicosapentaenoic Acid Versus Docosahexaenoic Acid on Serum Lipids: A Systematic Review and Meta-Analysis. Curr. Atheroscler. Rep. 2011, 13, 474–483. [Google Scholar] [CrossRef]
- Borow, K.M.; Nelson, J.R.; Mason, R.P. Biologic plausibility, cellular effects, and molecular mechanisms of eicosapentaenoic acid (EPA) in atherosclerosis. Atherosclerosis 2015, 242, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preston Mason, R. New Insights into Mechanisms of Action for Omega-3 Fatty Acids in Atherothrombotic Cardiovascular Disease. Curr. Atheroscler. Rep. 2019, 21, 2. [Google Scholar] [CrossRef] [PubMed]
- Drouin, G.; Guillocheau, E.; Catheline, D.; Baudry, C.; Le Ruyet, P.; Rioux, V.; Legrand, P. Impact of n-3 Docosapentaenoic Acid Supplementation on Fatty Acid Composition in Rat Differs Depending upon Tissues and Is Influenced by the Presence of Dairy Lipids in the Diet. J. Agric. Food Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.F.; Sinclair, A.J.; Kaur, G.; Li, D. Differential effects of EPA, DPA and DHA on cardio-metabolic risk factors in high-fat diet fed mice. Prostaglandins Leukot. Essent. Fat. Acids 2018, 66, 9976–9988. [Google Scholar] [CrossRef]
- Gotoh, N.; Nagao, K.; Onoda, S.; Shirouchi, B.; Furuya, R.; Nagai, T.; Mizobe, H.; Ichioka, K.; Watanabe, H.; Anagita, T.; et al. Effects of three different highly purified n-3 series highly unsaturated fatty acids on lipid metabolism in C57BL/KsJ-dbl db mice. J. Agric. Food Chem. 2009, 57, 11047–11054. [Google Scholar] [CrossRef]
- Gaetan, D.; Vincent, R.; Philippe, L. The n-3 docosapentaenoic acid (DPA): a new player in the n-3 long chain polyunsaturated fatty acid family. Biochimie 2019, 159, 36–48. [Google Scholar]
- Fialkow, J. Omega-3 fatty acid formulations in cardiovascular disease: Dietary supplements are not substitutes for prescription products. Am. J. Cardiovasc. Drugs 2016, 16, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, A.S.; Brown, T.J.; Brainard, J.S.; Biswas, P.; Thorpe, G.C.; Moore, H.J.; Deane, K.H.O.; AlAbdulghafoor, F.K.; Summerbell, C.D.; Worthington, H.V.; et al. Omega-3 fatty acids for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2018, 7, CD003177. [Google Scholar] [PubMed]
- Aung, T.; Halsey, J.; Kromhout, D.; Gerstein, H.C.; Marchioli, R.; Tavazzi, L.; Geleijnse, J.M.; Rauch, B.; Ness, A.; Galan, P.; et al. Associations of Omega-3 Fatty Acid Supplement Use With Cardiovascular Disease Risks. JAMA Cardiol. 2018, 3, 225. [Google Scholar] [CrossRef] [PubMed]
- The ASCEND Study Collaborative Group Effects of n−3 Fatty Acid Supplements in Diabetes Mellitus. N. Engl. J. Med. 2018, 379, 1540–1550. [CrossRef] [PubMed]
- Manson, J.E.; Cook, N.R.; Lee, I.-M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Albert, C.M.; Gordon, D.; Copeland, T.; et al. Marine n−3 Fatty Acids and Prevention of Cardiovascular Disease and Cancer. N. Engl. J. Med. 2019, 380, 23–32. [Google Scholar] [CrossRef]
- Michelle Kirkwood American Diabetes Association® Issues Critical Updates to the 2019 Standards of Medical Care in Diabetes. Available online: http://www.diabetes.org/newsroom/press-releases/2019/ada-issues-critical-updates-to-2019-standards-of-care.html (accessed on 9 May 2019).
- Kastelein, J.J.P.; Stroes, E.S.G. FISHing for the Miracle of Eicosapentaenoic Acid. N. Engl. J. Med. 2018, 380, 89–90. [Google Scholar] [CrossRef]
- Budoff, M.; Brent Muhlestein, J.; Le, V.T.; May, H.T.; Roy, S.; Nelson, J.R. Effect of Vascepa (icosapent ethyl) on progression of coronary atherosclerosis in patients with elevated triglycerides (200-499 mg/dL) on statin therapy: Rationale and design of the EVAPORATE study. Clin. Cardiol. 2018, 41, 13–19. [Google Scholar] [CrossRef]
- Matinas BioPharma Holdings I. Matinas BioPharma Reports 2018 Financial Results and Provides Corporate Update. Available online: https://www.matinasbiopharma.com/media/press-releases/detail/336/matinas-biopharma-reports-2018-financial-results-and (accessed on 7 May 2019).
- Stein, E. News-Novel PCSK9 inhibitor yields safe and effective LDL-c lowering. In Proceedings of the EAS 2019 Congress, Maastricht, The Netherlands, 26–29 May 2019. [Google Scholar]
- Van Poelgeest, E.P.; Hodges, M.R.; Moerland, M.; Tessier, Y.; Levin, A.A.; Persson, R.; Lindholm, M.W.; Dumong Erichsen, K.; Ørum, H.; Cohen, A.F.; et al. Antisense-mediated reduction of proprotein convertase subtilisin/kexin type 9 (PCSK9): A first-in-human randomized, placebo-controlled trial. Br. J. Clin. Pharmacol. 2015, 80, 1350–1361. [Google Scholar] [CrossRef]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef]
- Laufs, U.; Banach, M.; Mancini, G.B.J.; Gaudet, D.; Bloedon, L.T.; Sterling, L.R.; Kelly, S.; Stroes, E.S.G. Efficacy and Safety of Bempedoic Acid in Patients With Hypercholesterolemia and Statin Intolerance. J. Am. Heart Assoc. 2019, 8, e011662. [Google Scholar] [CrossRef] [Green Version]
- Catapano, A.L.; Tokgözoğlu, L.; Mello e Silva, A.; Bruckert, E. Pharmaceutical strategies for reducing LDL-C and risk of cardiovascular disease. Atheroscler. X 2019, 1, 100002. [Google Scholar] [CrossRef]
- Kingwell, B.A.; Chapman, M.J.; Kontush, A.; Miller, N.E. HDL-targeted therapies: progress, failures and future. Nat. Rev. Drug Discov. 2014, 13, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A.; Tsimikas, S. Lipid-Lowering Agents. Circ. Res. 2019, 124, 386–404. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.J.; Viney, N.; Crooke, R.M.; Tsimikas, S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J. Lipid Res. 2016, 57, 340–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toklu, B.; Amirian, J.; Robert, P.; Giugliano, F. Current Indications, Cost, and Clinical Use of Anti-PCSK9 Monoclonal Antibodies. Available online: https://www.acc.org/latest-in-cardiology/articles/2016/05/18/14/34/current-indications-cost-and-clinical-use-of-anti-pcsk9-monoclonal-antibodies (accessed on 10 May 2019).
- Ference, B.A.; Kastelein, J.J.; Ginsberg, H.N.; Chapman, M.J.; Nicholls, S.J.; Ray, K.K.; Packard, C.J.; Laufs, U.; Brook, R.D.; Oliver-Williams, C.; et al. Association of Triglyceride-Lowering LPL Variants and LDL-C–Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA 2019, 321, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Sniderman, A.D.; Pencina, M.; Thanassoulis, G. ApoB-The Power of Physiology to Transform the Prevention of Cardiovascular Disease. Circ. Res. 2019, 124, 1425–1427. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Name of the Drug | Mechanism of Action | Lipid Effect | Stage of Development | Comments* | |
|---|---|---|---|---|---|---|
| Lp(a) | 1. IONIS-APO(a) RX 2. IONIS-APO(a)-LRX | ASO to block apo(a) synthesis and thereby Lp(a) formation | 1. Lp(a) ↓ 72% Lp(a) ↓ 92% | [13] | Phase 2–3 | Lp(a) levels are not measured in the clinic |
| [14] | ||||||
| APOC-III | Volanesorsen (AKCEA-APOCIIIRX) | ASO to block ApoC-III synthesis | TG ↓ 73% | [15] | Phase 3 | Increases LDL cholesterol, thrombocytopenia risk |
| AKCEA-APOCIII-LRX | ASO to block ApoC-III synthesis | TG ↓ 71% | [16] | Phase 2 | ||
| ANGPTL3 | Evinacumab | Monoclonal antibodies target ANGPTL3 in plasma | TG ↓ 50% | [17] | Phase 3 | Reduces HDL cholesterol |
| IONIS-ANGPTL3-LRX | ASO to block ANGPTL3 synthesis | TG ↓ 50% | [18] | Phase 2 | Reduces HDL cholesterol | |
| OM3FA | 1. OM3FA-EE/Lovaza 2. OM3FA-CA/Epanova 3. OM3FA-IPE/Vascepa | Reduced TG levels, inflammation, oxidative properties of atherogenic lipoproteins and increases plaque instability | 1. TG ↓ 45% 2. TG ↓ 31% 3. TG ↓ 18–27% | [19,20] | Approved for HTG-patients | Inconclusive results on cardiovascular outcomes |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Zwol, W.; Rimbert, A.; Kuivenhoven, J.A. The Future of Lipid-Lowering Therapy. J. Clin. Med. 2019, 8, 1085. https://doi.org/10.3390/jcm8071085
van Zwol W, Rimbert A, Kuivenhoven JA. The Future of Lipid-Lowering Therapy. Journal of Clinical Medicine. 2019; 8(7):1085. https://doi.org/10.3390/jcm8071085
Chicago/Turabian Stylevan Zwol, Willemien, Antoine Rimbert, and Jan Albert Kuivenhoven. 2019. "The Future of Lipid-Lowering Therapy" Journal of Clinical Medicine 8, no. 7: 1085. https://doi.org/10.3390/jcm8071085